Amyloidosis¶
Chapter 117 | Harrison's 22e · Part 4 – Oncology: Hematologic Malignancies
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- See source text for full details
📑 Table of Contents¶
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🔀 Flowchart | Algorithm for the diagnosis of amyloidosis and determination of type |
| 1 | 🖼 Figure | Laboratory features of AL amyloidosis |
| 2 | 🖼 Figure | Laboratory features of AL amyloidosis |
| 3 | 🖼 Figure | Clinical signs of AL amyloidosis |
| 4 | 🖼 Figure | Clinical signs of AL amyloidosis |
| 5 | 🖼 Figure | Clinical signs of AL amyloidosis |
RAW CONTENT¶
[PAGE 895] Amyloidosis 895 CHAPTER 117 the assembly of light and heavy chains because they appear to contain AX, where A indicates amyloidosis and X represents the protein pres- both in their cytoplasm. Such patients are not treated differently from ent in the fibril. This chapter focuses primarily on the systemic forms. other patients with CLL (Chap. 107). AL amyloidosis refers to amyloid composed of immunoglobulin light chains; this disorder, formerly termed primary systemic amyloidosis, ■ FURTHER READING arises from a clonal B-cell or plasma cell disorder and can be associated Corre J et al: Risk factors in multiple myeloma: is it time for a revision? with myeloma or lymphoma. ATTR amyloidosis, the most prevalent Blood 137:16, 2021. of the familial amyloidoses, refers to amyloid derived from wild-type Hideshima T, Anderson KC: Signaling pathway mediating myeloma or mutated transthyretin (TTR), the transport protein for thyroid cell growth and survival. Cancers (Basel) 13:216, 2021. hormone and retinol-binding protein. AA amyloid is composed of the Hillengass J et al: International myeloma working group consensus acute-phase reactant protein serum amyloid A (SAA) and occurs in the recommendations on imaging in monoclonal plasma cell disorders. setting of chronic inflammatory or infectious diseases; for this reason, Lancet Oncol 20:e302, 2019. this type was formerly known as secondary amyloidosis. AβM amyloid Kumar S et al: International Myeloma Working Group consensus crite- results from misfolded β-microglobulin, occurring in indiv 2 iduals with ria for response and minimal residual disease assessment in multiple long-standing renal disea 2 se who have undergone dialysis, typically for myeloma. Lancet Oncol 17:e328, 2016. years. Aβ, the most common form of localized amyloidosis, is found Moreau P et al: Treatment of relapsed and refractory multiple in the brain of patients with Alzheimer’s disease after abnormal pro- myeloma: Recommendations from the International Myeloma Work- teolytic processing and aggregation of polypeptides derived from the ing Group. Lancet Oncol 22:e105, 2021. amyloid precursor protein. Munshi NC et al: A large meta-analysis establishes the role of MRD Diagnosis and treatment of the amyloidoses rest upon the histopath- negativity in long-term survival outcomes in patients with multiple ologic identification of amyloid deposits and immunohistochemical, myeloma. Blood Adv 4:5988, 2020. biochemical, or genetic determination of amyloid type (Fig. 117-1). Raje NS et al: Consensus guidelines and recommendations for infec- In the systemic amyloidoses, the clinically involved organs can be tion prevention in multiple myeloma: A report from the International biopsied, but amyloid deposits may be found in any tissue of the body. Myeloma Working Group. Lancet Haematol 9:e143 2022. Historically, blood vessels of the gingiva or rectal mucosa were often Rajkumar SV et al: International Myeloma Working Group updated examined, but the most easily accessible tissue—positive in more than criteria for the diagnosis of multiple myeloma. Lancet Oncol 15:e538, 80% of patients with systemic amyloidosis—is abdominal fat. After 2014. local anesthesia, fat is aspirated with a 16-gauge needle from the subcu- Richardson PG et al: Triplet therapy, transplantation, and mainte- taneous layer of the abdominal wall. Fat globules expelled onto a glass nance until progression in myeloma. N Engl J Med 387:132, 2022. slide can be stained for amyloid by Congo red dye, thus avoiding a sur- Robiou du Pont S et al: Genomics of multiple myeloma. J Clin Oncol gical procedure. If this material is negative, more invasive biopsies of 35:963, 2017. the involved organ like kidney, heart, liver, tongue, or gastrointestinal Terpos E et al: Treatment of multiple myeloma-related bone disease: tract can be considered in patients in whom amyloidosis is suspected. Recommendations from the Bone Working Group of the Interna- The regular β-sheet structure of amyloid deposits exhibits a unique tional Myeloma Working Group. Lancet Oncol 22:e119, 2021. “green” birefringence by polarized light microscopy when stained with Treon SP et al: How I use genomics and BTK inhibitors in the treat- Congo red dye; other regular protein structures (e.g., collagen) appear ment of Waldenstrom macroglobulinemia. Blood 143:1702, 2024. white under these conditions. The 10-nm-diameter fibrils can also be visualized by electron microscopy of paraformaldehyde-fixed tissue. Once amyloid is found, the precursor protein type must be determined by immunohistochemistry, immunoelectron microscopy, or extraction and biochemical analysis employing mass spectrometry; gene sequenc- ing is used to identify mutants causing hereditary amyloidosis. How- ever, a mass spectrometry–based analysis of the amyloid-containing 117 Amyloidosis tissues is now considered the best approach, with a reported sensitivity of 88% and specificity of 96%, which are higher than immunochemical John L. Berk, Vaishali Sanchorawala techniques, and this technique does not require a large panel of antisera to identify non-AL amyloidosis. The patient’s history, physical find- ings, and clinical presentation, including age and ethnic origin, organ ■ GENERAL PRINCIPLES system involvement, underlying diseases, and family history, may pro- Amyloidosis is the term for a group of protein misfolding disorders vide helpful clues as to the type of amyloidosis. However, there can be characterized by the extracellular deposition of insoluble polymeric considerable overlap in clinical presentations, and accurate typing of protein fibrils in tissues and organs. A robust cellular machinery exists the amyloidogenic protein is essential to guide appropriate therapy and to chaperone proteins during the process of synthesis and secretion, to offer genetic counseling as appropriate. ensure that they achieve correct tertiary conformation and function, The mechanisms of fibril formation and tissue toxicity remain con- and to eliminate proteins that misfold. However, genetic mutation, troversial. The “amyloid hypothesis,” as it is currently understood, pro- incorrect processing, and other factors may favor misfolding, with poses that precursor proteins undergo a process of reversible unfolding consequent loss of normal protein function and intracellular or extra- or misfolding; misfolded proteins form oligomeric aggregates, higher- cellular aggregation. Many diseases, ranging from cystic fibrosis to order polymers, and then fibrils that deposit in tissues. Accumulating Alzheimer’s disease, are now known to involve protein misfolding. evidence suggests that the oligomeric intermediates may constitute the In the amyloidoses, the aggregates are typically extracellular, and the most toxic species. Oligomers are more capable than fibrils of interact- misfolded protein subunits assume a common antiparallel, β-pleated ing with cells and inducing formation of reactive oxygen species and sheet–rich structural conformation that leads to the formation of stress signaling. Ultimately, the fibrillar tissue deposits are likely to higher-order oligomers and then fibrils with unique staining proper- interfere with normal organ function. However, direct proteotoxicity ties. The term amyloid was coined around 1854 by the pathologist of the soluble oligomers also can lead to organ dysfunction. A more Rudolf Virchow, who thought that these deposits resembled starch sophisticated understanding of the mechanisms leading to amyloid (Latin amylum) under the microscope. formation and cell and tissue dysfunction will continue to provide new Amyloid diseases, defined by the biochemical nature of the protein targets for therapies. composing the fibril deposits, are classified according to whether they The clinical syndromes of the amyloidoses are associated with rela- are systemic or localized, whether they are acquired or inherited, and tively nonspecific alterations in routine laboratory t
Flowcharts & Algorithms¶
Reproduced from Harrison's 22nd Edition.
Flowchart 1¶

Caption: FIGURE 117-1 Algorithm for the diagnosis of amyloidosis and determination of type. unexplained nephropathy, cardiomyopathy, neuropathy, enteropathy, arthropathy, and apolipoprotein AI; ApoAII, apolipoprotein AII; GI, gastrointestinal; IHC,
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶
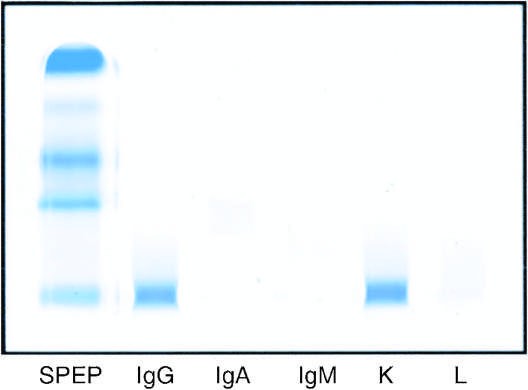
Caption: FIGURE 117-3 Laboratory features of AL amyloidosis. A. Serum immunofixation electrophoresis reveals an IgGκ monoclonal protein in this example; serum protein electrophoresis is often normal. B. Bone marrow biopsy sections stained by immunohistochemistry with antibody to CD138 (syndecan, highly expressed on plasma cells) (left) or by in situ hybridization with fluorescein-tagged probes (Ventana Medical Systems) binding to κ mRNA (center) and λ mRNA (right) in plasma cells. (Photomicrograph courtesy of C. O’Hara; with permission.)
Figure 2¶
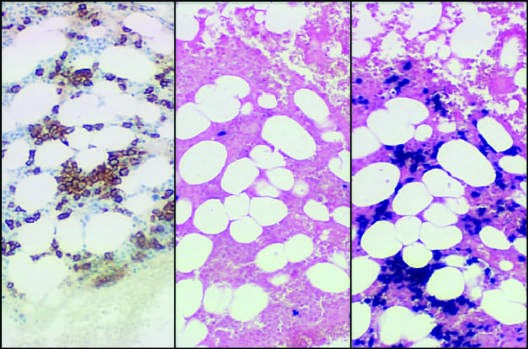
Caption: FIGURE 117-3 Laboratory features of AL amyloidosis. A. Serum immunofixation electrophoresis reveals an IgGκ monoclonal protein in this example; serum protein electrophoresis is often normal. B. Bone marrow biopsy sections stained by immunohistochemistry with antibody to CD138 (syndecan, highly expressed on plasma cells) (left) or by in situ hybridization with fluorescein-tagged probes (Ventana Medical Systems) binding to κ mRNA (center) and λ mRNA (right) in plasma cells. (Photomicrograph courtesy of C. O’Hara; with permission.)
Figure 3¶
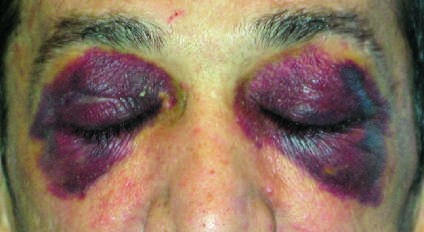
Caption: FIGURE 117-2 Clinical signs of AL amyloidosis. A. Macroglossia. B. Periorbital ecchymoses. C. Fingernail dystrophy.
Figure 4¶
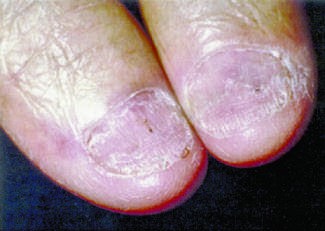
Caption: FIGURE 117-2 Clinical signs of AL amyloidosis. A. Macroglossia. B. Periorbital ecchymoses. C. Fingernail dystrophy.
Figure 5¶
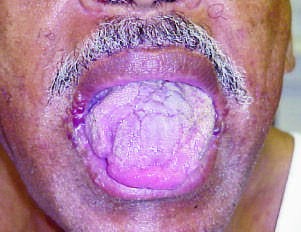
Caption: FIGURE 117-2 Clinical signs of AL amyloidosis. A. Macroglossia. B. Periorbital ecchymoses. C. Fingernail dystrophy.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.