Chapter 399: Pheochromocytoma¶
Chapter 399 | Part 12: Endocrinology · Part 12 – Endocrinology & Metabolism
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- Pheochromocytomas (PPGLs) are catecholamine-producing tumors derived from the sympathetic or parasympathetic nervous system.
- The classic 'rule of tens' states that ~10% are bilateral, 10% are extra-adrenal, and 10% are metastatic.
- Approximately 25–33% of patients have an inherited condition (germline mutations in RET, VHL, NF1, SDHx, etc.).
- The classic triad of palpitations, headache, and profuse sweating is seen in roughly a third of patients.
- Diagnosis requires biochemical testing (plasma/urinary metanephrines) and imaging (CT/MRI).
- Preoperative alpha-adrenergic blockade (e.g., phenoxybenzamine) is mandatory before beta-blockade to prevent hypertensive crisis.
- Universal germline panel testing is the gold standard for genetic characterization of PPGLs.
- Metastatic pheochromocytoma is defined by distant metastases (lungs, bone, liver), not just histologic atypia.
- MEN 2 syndromes (RET mutations) are associated with medullary thyroid carcinoma and pheochromocytoma.
- VHL disease is associated with retinal/cerebellar hemangioblastomas and renal cell carcinoma.
- Surgery is the ultimate therapeutic goal, performed by experienced teams with minimally invasive techniques.
- 5-year survival rates for metastatic disease range from 30–60%.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 Terminology & Classification
- 2. EPIDEMIOLOGY
- 2.1 The Rule of Tens
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Genetic Syndromes
- 4. CLINICAL FEATURES
- 4.1 Clinical Features Table
- 4.2 Precipitants of Crisis
- 5. DIFFERENTIAL DIAGNOSIS
- 5.1 Mimickers
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Biochemical Testing Sensitivity & Specificity
- 6.2 Diagnostic Algorithm
- 7. MANAGEMENT & TREATMENT
- 7.1 Preoperative Preparation
- 7.2 Surgical Management
- 7.3 Metastatic Disease Treatment
- 8. PROGNOSIS & COMPLICATIONS
- 8.1 Malignancy Criteria
- 9. SPECIAL CONSIDERATIONS
- 9.1 Pregnancy
- 9.2 Genetic Screening
- 10. KEY PEARLS & CLINICAL TRAPS
- WHAT TO LOOK FOR — DIAGNOSTIC CLUES
- WHAT EXCLUDES THE DIAGNOSIS
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🖼 Figure | The paraganglial system and topographic sites (in red) of Manger, RW Gifford:... |
| 2 | 🖼 Figure | von Hippel–Lindau disease |
| 3 | 🖼 Figure | Typical pheochromocytoma (adrenal unilateral) |
| 4 | 🖼 Figure | Multiple and metastatic pheochromocytoma |
| 5 | 🖼 Figure | Multiple and metastatic pheochromocytoma |
| 6 | 🖼 Figure | Typical pheochromocytoma (adrenal unilateral) |
| 7 | 🖼 Figure | Multiple and metastatic pheochromocytoma |
| 8 | 🖼 Figure | Multiple and metastatic pheochromocytoma |
| 9 | 🖼 Figure | Multiple and metastatic pheochromocytoma |
| 10 | 🖼 Figure | Multiple and metastatic pheochromocytoma |
| 11 | 🖼 Figure | Multiple and metastatic pheochromocytoma |
| 12 | 🖼 Figure | Typical pheochromocytoma (adrenal unilateral) |
| 13 | 🖼 Figure | Multiple and metastatic pheochromocytoma |
| 14 | 🖼 Figure | Typical pheochromocytoma (adrenal unilateral) |
1. DEFINITION & OVERVIEW¶
- Pheochromocytomas and paragangliomas (PPGLs) are catecholamine-producing tumors derived from the sympathetic or parasympathetic nervous system.
- These tumors may arise sporadically or be inherited as features of multiple endocrine neoplasia type 2 (MEN 2), von Hippel–Lindau (VHL) disease, or several other pheochromocytoma-associated syndromes.
- The diagnosis of pheochromocytomas identifies a potentially correctable cause of hypertension, and their removal can prevent hypertensive crises that can be lethal.
- The clinical presentation is variable, ranging from an adrenal incidentaloma to a hypertensive crisis with associated cerebrovascular or cardiac complications.
- Harrison's defines this as: 'The name pheochromocytoma reflects the formerly used black-colored staining caused by chromaffin oxidation of catecholamines.'
- The World Health Organization (WHO) applies the term pheochromocytoma to adrenal tumors (usually secreting) and the term paraganglioma to tumors at all other sites including head and neck, thoracic, extra-adrenal retroperitoneal, and pelvic sites.
1.1 Terminology & Classification¶
- Pheochromocytoma: Adrenal tumors (usually secreting).
- Paraganglioma: Tumors at all other sites including head and neck, thoracic, extra-adrenal retroperitoneal, and pelvic sites.
- PPGLs are well-vascularized tumors that arise from cells derived from the sympathetic (e.g., adrenal medulla or sympathetic trunk) or parasympathetic (e.g., carotid body, glomus tympanicum, glomus jugulare, glomus vagale) paraganglia.
2. EPIDEMIOLOGY¶
- The incidence of PPGL ranges from 0.04 to 0.95 cases per 100,000 per year.
- There is a gradual increase over time probably due to genetic familial screening, changes in imaging modalities, and more frequent detection.
- About ~0.1% of hypertensive patients harbor a pheochromocytoma.
- The mean age at diagnosis is ~40 years, although the tumors can be detected from early childhood, particularly when genetically determined, until late in life.
- At diagnosis, patients with inherited syndromes are a mean of ~15 years younger than patients with sporadic tumors.
2.1 The Rule of Tens¶
- The classic 'rule of tens' for pheochromocytomas states that ~10% are bilateral.
- ~10% are extra-adrenal.
- ~10% are metastatic.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- The etiology of sporadic PPGLs is unknown.
- However, 25–33% of patients have an inherited condition, including germline mutations in the classically recognized RET, VHL, NF1, SDHB, SDHC, and SDHD genes or in the more recently recognized SDHA, SDHAF2, TMEM127, MAX, FH, PDH1, PDH2, HIF1α, HIF2α, MDH2, KIF1Bβ, IDH1, SLC25A11, H-RAS, and DNMTA3 genes.
- Biallelic gene inactivation, a characteristic of tumor-suppressor genes, has been demonstrated for the VHL, NF1, SDHx, TMEM127, MAX, FH, PDH1, PDH2, MDH2, and KIF1Bβ genes.
- In contrast, RET is a protooncogene, and mutations activate receptor tyrosine kinase activity.
- Succinate dehydrogenase (SDH) is an enzyme of the Krebs cycle and the mitochondrial respiratory chain.
- The VHL protein is a component of a ubiquitin E3 ligase. VHL mutations reduce protein degradation, resulting in upregulation of components involved in cell-cycle progression, glucose metabolism, and oxygen sensing.
- Pathogenetically relevant genes are broadly grouped into three different clusters: cluster 1 (pseudohypoxia group: SDHx, FH, VHL, HIF2A), cluster 2 (kinase signaling group: RET, NF1, TMEM127, MAX, HRAS, KIF1Bβ, PDH), and cluster 3 (Wnt signaling group: CSDE1, MAML3).
3.1 Genetic Syndromes¶
- MEN 2 (Multiple Endocrine Neoplasia type 2): Caused by mutations in RET, which encodes a tyrosine kinase. Both types (2A and 2B) are autosomal dominant.
- MEN 2A: Characterized by medullary thyroid carcinoma (MTC), pheochromocytoma, and hyperparathyroidism.
- MEN 2B: Also includes MTC (more aggressive), pheochromocytoma, and multiple mucosal neuromas, marfanoid habitus, and other developmental disorders, although it typically lacks hyperparathyroidism.
- VHL (von Hippel–Lindau): Autosomal dominant disorder predisposing to retinal and cerebellar hemangioblastomas, clear cell renal carcinomas, pancreatic neuroendocrine tumors, and other tumors.
- NF1 (Neurofibromatosis type 1): The NF1 gene functions as a tumor suppressor by regulating the Ras signaling cascade. Classic features include multiple neurofibromas, café au lait spots, axillary freckling, and Lisch nodules.
- PGL Syndromes (Paraganglioma Syndromes): Classified by genetic analyses of families with head and neck paragangliomas. Susceptibility genes encode subunits of the enzyme SDH (SDHA, SDHB, SDHC, SDHD, SDHAF2).
- TMEM127 and MAX: Hereditary mainly adrenal tumors in patients with germline mutations in these genes.
- Pituitary neuroendocrine tumors: Described as an association with SDHx (pituitary adenoma and pheochromocytoma/paraganglioma 3PA syndrome) as well as MAX mutations (MEN 5), but they seem to occur very rarely.
4. CLINICAL FEATURES¶
- Its clinical presentation is so variable that pheochromocytoma has been termed 'the great masquerader'.
- Among the presenting manifestations, episodes of palpitation, headache, and profuse sweating are typical, and these manifestations constitute a classic triad that is seen in roughly a third of patients with pheochromocytoma.
- The presence of all three manifestations in association with hypertension makes pheochromocytoma a likely diagnosis.
- However, a pheochromocytoma can be asymptomatic for years or can be identified through imaging screening in a patient presenting with a hereditary syndrome.
- Some tumors grow to a considerable size before patients note symptoms.
- The dominant sign is hypertension. Classically, patients have episodic hypertension, but sustained hypertension is also common.
- Catecholamine crises can lead to heart failure, pulmonary edema, arrhythmias, and intracranial hemorrhage.
- During episodes of hormone release, which can occur at widely divergent intervals, patients are anxious and pale, and they experience tachycardia and palpitations.
- These paroxysms generally last <1 h and may be precipitated by surgery, positional changes, exercise, pregnancy, urination (particularly with bladder pheochromocytomas), and various medications.
- Patients may present with true panic attacks that may be mistakenly attributed to psychiatric illness.
4.1 Clinical Features Table¶
- Headaches
- Profuse sweating
- Palpitations and tachycardia
- Hypertension, sustained or paroxysmal
- Anxiety and panic attacks
- Pallor
- Nausea
- Abdominal pain
- Weakness
- Weight loss
- Paradoxical response to antihypertensive drugs
- Polyuria and polydipsia
- Constipation
- Orthostatic hypotension
- Dilated cardiomyopathy
- Erythrocytosis
- Elevated blood sugar
- Hypercalcemia
4.2 Precipitants of Crisis¶
- Surgery
- Positional changes
- Exercise
- Pregnancy
- Urination (particularly with bladder pheochromocytomas)
- Various medications (e.g., tricyclic antidepressants, opiates, metoclopramide)
5. DIFFERENTIAL DIAGNOSIS¶
- When the possibility of a pheochromocytoma is being entertained, other disorders to consider include essential hypertension, anxiety attacks, use of cocaine or amphetamines, mastocytosis or carcinoid syndrome (usually without hypertension), intracranial lesions, clonidine withdrawal, autonomic epilepsy, and factitious crises (usually from use of sympathomimetic amines).
- When an asymptomatic adrenal mass is identified, likely diagnoses other than pheochromocytoma include a nonfunctioning adrenal adenoma, an aldosteronoma, and a cortisol-producing adenoma (Cushing's syndrome).
5.1 Mimickers¶
- Essential hypertension
- Anxiety attacks
- Use of cocaine or amphetamines
- Mastocytosis or carcinoid syndrome (usually without hypertension)
- Intracranial lesions
- Clonidine withdrawal
- Autonomic epilepsy
- Factitious crises (usually from use of sympathomimetic amines)
6. INVESTIGATIONS & DIAGNOSIS¶
- The diagnosis is based on documentation of catecholamine excess by biochemical testing and localization of the tumor by imaging.
- These two criteria are of equal importance, although measurement of catecholamines or metanephrines (their methylated metabolites) is traditionally the first step in diagnosis.
- CT or MRI, prove to be pheochromocytomas upon endocrinologic evaluation, but the presence of pheochromocytomas is unlikely if unenhanced CT reveals an attenuation of 95%).
- These agents are particularly useful in the documentation of hereditary syndromes but also in metastatic pheochromocytoma, because uptake is exhibited also in paragangliomas and metastases.
- Pathology: PPGLs are found at the classical sites of the adrenal medulla and paraganglia.
- Histologically, the tumors often show a characteristic 'Zellballen' pattern, consisting of nests of neuroendocrine chief cells with peripheral glial-like sustentacular cells.
- However, a broad spectrum of architectural and cytologic features can be seen.
- Immunohistochemistry is positive for chromogranin and synaptophysin in the chief cells and S-100 in the sustentacular cells.
- Increasingly, staining with antibodies against the proteins encoded by susceptibility genes for hereditary pheochromocytomas, such as SDHB, is used to histologically demonstrate defects of these proteins, thereby making germline mutations more likely.
6.1 Biochemical Testing Sensitivity & Specificity¶
- 24-h urinary tests:
- Catecholamines: Sensitivity +++ (85–95%), Specificity +++ (85–95%).
- Fractionated metanephrines: Sensitivity ++ (85–95%), Specificity ++ (95%).
- Plasma tests:
- Catecholamines: Sensitivity +++ (85–95%), Specificity ++ (95%), Specificity +++ (85–95%).
- Imaging:
- CT: Sensitivity ++++ (>95%), Specificity +++ (85–95%).
- MRI: Sensitivity ++++ (>95%), Specificity +++ (85–95%).
- MIBG scintigraphy: Sensitivity ++ (85–95%), Specificity ++++ (>95%).
- Somatostatin receptor scintigraphy: Sensitivity ++ (85–95%), Specificity ++ (85–95%).
- 68Gallium-DOTATOC or DOTATATE PET/CT: Sensitivity ++++ (>95%), Specificity ++++ (>95%).
- 18Fluoro-DOPA PET/CT: Sensitivity ++++ (>95%), Specificity ++ (85–95%).
- 68Gallium-DOTATOC or DOTATATE PET/CT: Sensitivity ++++ (>95%), Specificity ++++ (>95%).
6.2 Diagnostic Algorithm¶
- Step 1: Suspect pheochromocytoma based on clinical presentation (headache, palpitations, sweating, hypertension).
- Step 2: Perform biochemical testing (plasma or urinary metanephrines and catecholamines).
- Step 3: If biochemical testing is positive, perform imaging (CT or MRI).
- Step 4: If imaging is negative but biochemical testing is positive, consider repeat testing or alternative imaging (MIBG, PET).
- Step 5: If biochemical testing is borderline, exclude dietary or drug-related factors (withdrawal of levodopa or use of sympathomimetics, diuretics, tricyclic antidepressants, opiates, metoclopramide, alpha and beta blockers).
- Step 6: If biochemical testing is three times the upper limit of normal, diagnosis is highly likely regardless of the assay used.
7. MANAGEMENT & TREATMENT¶
- Complete tumor removal, the ultimate therapeutic goal, can be used by partial or total adrenalectomy.
- It is important to preserve the normal adrenal cortex in order to prevent Addison's disease, particularly in hereditary disorders in which bilateral pheochromocytomas are most likely.
- Preoperative preparation of the patient has to be considered, and blood pressure should be consistently <160/90 mmHg.
- Classically, blood pressure has been controlled by α-adrenergic blockers (oral phenoxybenzamine, 0.5–4 mg/kg of body weight).
- Because patients are volume-constricted, liberal salt intake and hydration are necessary to avoid severe orthostasis.
- Oral prazosin or intravenous phentolamine can be used to manage paroxysms while adequate alpha blockade is awaited.
- Beta blockers (e.g., 10 mg of propranolol three or four times per day) should not be used as first-line treatment because of the risk of increased hypertension.
- Other antihypertensives, such as calcium channel blockers or angiotensin-converting enzyme inhibitors, have also been used effectively.
- Surgery should be performed by teams of surgeons and anesthesiologists with experience in the management of pheochromocytomas.
- Blood pressure can be labile during surgery, particularly at the outset of intubation or when the tumor is manipulated.
- Nitroprusside infusion is useful for intraoperative hypertensive crises, and hypotension usually responds to volume infusion.
- The latter side effect can, however, be avoided in normotensive pheochromocytoma patients by having only standby intraoperative nitroprusside, which has been shown to be safe and avoids postoperative hypotension often caused by alpha blockers.
- The long-lasting guideline for obligatory preoperative treatment with alpha blockers is under discussion and seemingly not needed.
- Minimally invasive techniques (laparoscopy or retroperitoneoscopy) have become the standard approaches in pheochromocytoma surgery.
- They are associated with fewer complications, a faster recovery, and optimal cosmetic results.
- Extra-adrenal abdominal and thoracic pheochromocytomas can also be removed endoscopically.
- In this setting, adrenal sparing surgery should be considered.
- Postoperatively, catecholamine normalization should be documented.
- An adrenocorticotropic hormone (ACTH) test should be used to exclude cortisol deficiency when bilateral adrenal cortex–sparing surgery has been performed.
- Head and neck paragangliomas are a challenge for surgeons, since damage of adjacent tissue, mainly vessels or cranial nerves II, VII, IX, X, XI, and XII, is a frequent permanent side effect.
- Careful consideration of best management is important, and radiotherapy may be an alternative, especially for large head and neck paragangliomas.
- Tympanic paragangliomas are symptomatic early, and most of these tumors can easily be resected, with subsequent improvement of hearing and alleviation of tinnitus.
- Asymptomatic paraganglial tumors, now often detected in patients with hereditary tumors and their relatives, are challenging to manage.
- Watchful waiting strategies have been introduced, but they should consider any genetic syndrome, such as SDHB, that might be associated with a higher degree of malignancy.
- Treatment of metastatic pheochromocytoma or paraganglioma is challenging.
- Options include tumor mass reduction, alpha blockers for symptoms, chemotherapy including tyrosine kinase inhibitors, nuclear medicine radiotherapy, and stereotactic radiation.
- Nuclear medicine therapy is the treatment of choice for scintigraphically documented metastases, preferably with 131I-MIBG in 100–300 mCi doses over 3–6 cycles, or somatostatin receptor ligands, e.g., DOTATOC labeled with yttrium-90 or lutetium-177.
- Averbuch's chemotherapy protocol includes dacarbazine (600 mg/m2 on days 1 and 2), cyclophosphamide (750 mg/m2 on day 1), and vincristine (1.4 mg/m2 on day 1), all repeated every 21 days for 3–6 cycles.
- Palliation (stable disease to shrinkage) is achieved in about one-half of patients.
- Due to increasing insights in the genetics of pheochromocytoma and their molecular pathways, new targeted chemotherapeutic options such as sunitinib and temozolomide are under investigation.
7.1 Preoperative Preparation¶
- Blood pressure target: <160/90 mmHg.
- Alpha-adrenergic blockers: Oral phenoxybenzamine, 0.5–4 mg/kg of body weight.
- Salt intake: Liberal salt intake and hydration are necessary to avoid severe orthostasis.
- Beta blockers: Should not be used as first-line treatment because of the risk of increased hypertension.
- Other antihypertensives: Calcium channel blockers or angiotensin-converting enzyme inhibitors have also been used effectively.
- Oral prazosin or intravenous phentolamine: Can be used to manage paroxysms while adequate alpha blockade is awaited.
7.2 Surgical Management¶
- Technique: Minimally invasive techniques (laparoscopy or retroperitoneoscopy) have become the standard approaches.
- Benefits: Associated with fewer complications, a faster recovery, and optimal cosmetic results.
- Extra-adrenal tumors: Can also be removed endoscopically.
- Adrenal sparing: Should be considered in this setting.
- Intraoperative management: Nitroprusside infusion is useful for intraoperative hypertensive crises.
- Hypotension management: Volume infusion usually responds to hypotension.
- Standby nitroprusside: Avoids postoperative hypotension often caused by alpha blockers.
7.3 Metastatic Disease Treatment¶
- Nuclear medicine therapy: Treatment of choice for scintigraphically documented metastases.
- 131I-MIBG: 100–300 mCi doses over 3–6 cycles.
- Somatostatin receptor ligands: e.g., DOTATOC labeled with yttrium-90 or lutetium-177.
- Chemotherapy: Averbuch's protocol (dacarbazine, cyclophosphamide, vincristine).
- Targeted therapy: Sunitinib and temozolomide are under investigation.
8. PROGNOSIS & COMPLICATIONS¶
- About 5–10% of PPGLs are metastatic.
- The diagnosis of malignant pheochromocytoma is problematic.
- The typical histologic criteria of cellular atypia, presence of mitoses, and invasion of vessels or adjacent tissues are insufficient for the diagnosis of malignancy in pheochromocytoma.
- Size >5 cm, high pheochromocytoma of the adrenal gland scaled score (PASS) and grading system for adrenal pheochromocytoma and paraganglioma (GAPP) score, and SDHB-positive status have been considered as markers of risk of recurrence, but they remain controversial.
- Thus, the term malignant pheochromocytoma has been replaced by metastatic pheochromocytoma as suggested by the WHO and is restricted to tumors with lymph node or distant metastases, the latter most commonly found by nuclear medicine imaging in lungs, bone, or liver locations, suggesting a vascular pathway of spread.
- Because hereditary syndromes are associated with multifocal tumor sites, these features should be anticipated in patients with germline mutations, especially of SDHB, SDHD, VHL, and RET.
- However, distant metastases also occur in these syndromes, especially in carriers of SDHB mutations.
- The prognosis of metastatic pheochromocytoma or paraganglioma is variable, with 5-year survival rates of 30–60%.
8.1 Malignancy Criteria¶
- Cellular atypia
- Presence of mitoses
- Invasion of vessels or adjacent tissues
- Size >5 cm
- High pheochromocytoma of the adrenal gland scaled score (PASS)
- Grading system for adrenal pheochromocytoma and paraganglioma (GAPP) score
- SDHB-positive status
- Distant metastases (lungs, bone, liver)
9. SPECIAL CONSIDERATIONS¶
- Pheochromocytomas occasionally are diagnosed in pregnancy and can be very challenging to manage.
- The pathogenesis of adrenergic crises during pregnancy in previously asymptomatic women might be linked to human chorionic gonadotropin (hCG)-induced stimulation of epinephrine production by pheochromocytoma, as some of these express luteinizing hormone/chorionic gonadotropin (LHCG) receptors.
- Endoscopic removal, preferably in the fourth to sixth month of gestation, is possible and can be followed by uneventful childbirth.
- Regular screening in families with inherited pheochromocytomas provides an opportunity to identify and remove such tumors in women of most reproductive age.
- Head and neck paragangliomas are a challenge for surgeons, since damage of adjacent tissue, mainly vessels or cranial nerves II, VII, IX, X, XI, and XII, is a frequent permanent side effect.
- Careful consideration of best management is important, and radiotherapy may be an alternative, especially for large head and neck paragangliomas.
- Tympanic paragangliomas are symptomatic early, and most of these tumors can easily be resected, with subsequent improvement of hearing and alleviation of tinnitus.
- Asymptomatic paraganglial tumors, now often detected in patients with hereditary tumors and their relatives, are challenging to manage.
- Watchful waiting strategies have been introduced, but they should consider any genetic syndrome, such as SDHB, that might be associated with a higher degree of malignancy.
9.1 Pregnancy¶
- Diagnosis: Occasionally diagnosed in pregnancy.
- Pathogenesis: hCG-induced stimulation of epinephrine production.
- Management: Endoscopic removal, preferably in the fourth to sixth month of gestation.
- Outcome: Can be followed by uneventful childbirth.
9.2 Genetic Screening¶
- Universal germline panel testing is now the gold standard to characterize the genetic factors involved in PPGL.
- It usually identifies a genetic etiology in up to 30% of the cases.
- This rate can be even higher in patients with early age of onset, extra-adrenal location, multiple tumors, metastatic tumors, or family history of PPGL.
- Effective preventive medicine for pheochromocytoma and pheochromocytoma-associated diseases requires management according to identified germline mutations in susceptibility genes.
- Because of the relatively high prevalence of familial syndromes among patients who present with pheochromocytoma or paraganglioma, it is useful to identify germline mutations even in patients without a known family history.
- Despite the use of a universal germline panel testing performed for all patients with a PPGL, a first step remains to search for clinical features of inherited syndromes and to obtain an in-depth, multigenerational family history.
- Each of these syndromes exhibits autosomal dominant transmission with variable penetrance, but a proband with a mother affected by paraganglial tumors is not predisposed to PGL1 and PGL2 (SDHD and SDHAF2 mutation carrier).
- Cutaneous neurofibromas, café au lait spots, and axillary freckling suggest neurofibromatosis.
- Germline mutations in NF1 have nearly never been reported in patients with sporadic pheochromocytomas. Thus, NF1 testing is not needed in the absence of other clinical features of neurofibromatosis.
- A personal or family history of MTC or an elevation of serum calcitonin strongly suggests MEN 2 and should prompt testing for RET mutations.
- A history of visual impairment or tumors of the cerebellum, brainstem, spinal cord, or the kidney suggests the possibility of VHL.
- A personal and/or family history of head and neck paraganglioma suggests PGL1 or PGL4.
- Of note, sequencing protocols may not detect large deletions of one or more exons.
10. KEY PEARLS & CLINICAL TRAPS¶
- The classic 'rule of tens' states that ~10% are bilateral, 10% are extra-adrenal, and 10% are metastatic.
- The presence of all three manifestations (palpitations, headache, sweating) in association with hypertension makes pheochromocytoma a likely diagnosis.
- Beta blockers should not be used as first-line treatment because of the risk of increased hypertension.
- Preoperative alpha-adrenergic blockade is mandatory before beta-blockade.
- Universal germline panel testing is the gold standard for genetic characterization.
- Metastatic pheochromocytoma is defined by distant metastases, not just histologic atypia.
- About 5% of adrenal incidentalomas prove to be pheochromocytomas upon endocrinologic evaluation.
- The presence of pheochromocytomas is unlikely if unenhanced CT reveals an attenuation of <10 Hounsfield units (HU).
- Head and neck paragangliomas are a challenge for surgeons due to damage of adjacent tissue.
- Watchful waiting strategies should consider any genetic syndrome, such as SDHB, that might be associated with a higher degree of malignancy.
WHAT TO LOOK FOR — DIAGNOSTIC CLUES¶
- Classic triad: Palpitations, headache, and profuse sweating seen in roughly a third of patients.
- Dominant sign: Hypertension (episodic or sustained).
- Characteristic fluctuations in the hormonal activity of tumors result in considerable variation in serial catecholamine measurements.
- Most tumors continuously leak O-methylated metabolites, which are detected by measurement of metanephrines.
- When pheochromocytoma is suspected on clinical grounds (i.e., when values are three times the upper limit of normal), this diagnosis is highly likely regardless of the assay used.
- For PET-CT with both 68Ga-DOTATATE and 18F-DOPA, the sensitivity and specificity are very high (>95%).
- These agents are particularly useful in the documentation of hereditary syndromes but also in metastatic pheochromocytoma, because uptake is exhibited also in paragangliomas and metastases.
- T2-weighted MRI with gadolinium contrast is optimal for detecting pheochromocytomas and is somewhat better than CT for imaging extra-adrenal PPGLs.
- Histologically, the tumors often show a characteristic 'Zellballen' pattern, consisting of nests of neuroendocrine chief cells with peripheral glial-like sustentacular cells.
- Immunohistochemistry is positive for chromogranin and synaptophysin in the chief cells and S-100 in the sustentacular cells.
- Increasingly, staining with antibodies against the proteins encoded by susceptibility genes for hereditary pheochromocytomas, such as SDHB, is used to histologically demonstrate defects of these proteins, thereby making germline mutations more likely.
WHAT EXCLUDES THE DIAGNOSIS¶
- The presence of pheochromocytomas is unlikely if unenhanced CT reveals an attenuation of <10 Hounsfield units (HU).
- Borderline elevations are likely to represent false-positive results.
- In this circumstance, it is important to exclude dietary or drug-related factors (withdrawal of levodopa or use of sympathomimetics, diuretics, tricyclic antidepressants, opiates, metoclopramide, alpha and beta blockers) that might cause false-positive results and then to repeat testing.
- The typical histologic criteria of cellular atypia, presence of mitoses, and invasion of vessels or adjacent tissues are insufficient for the diagnosis of malignancy in pheochromocytoma.
- Distant metastases also occur in these syndromes, especially in carriers of SDHB mutations, but the diagnosis of malignant pheochromocytoma is problematic and restricted to tumors with lymph node or distant metastases.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 399-1 The paraganglial system and topographic sites (in red) of Manger, RW Gifford: Clinical and experimental pheochromocytoma. Cambridge: — Figure 399-1: The paraganglial system and topographic sites (in red) of pheochromocytomas and paragangliomas, including adrenal, extra-adrenal, and head and neck locations.
Figure 2¶

Caption: FIGURE 399-6 von Hippel–Lindau disease. Tumors and cysts marked by arrows. A. findings on magnetic resonance imaging. B–D. Hemangioblastomas of the cerebellum cord (thoracic) (D). E. Bilateral renal clear cell carcinomas with two tumors on each side multiple tiny spaces). H. Two pancreatic islet cell tumors. (Part A was provided Freiburg. Part C was provided courtesy of Dr. Sven Glaesker, Brussels. Part D was used of Dr. Cordula Jilg, Freiburg. Parts F–H were provided courtesy of Dr. Tobias Krauss, SDHA, SDHB, and SDHC germline mutations is autosomal dominant. in In contrast, in virtually all SDHD and SDHAF2 families, only the prog- eny of affected fathers develops tumors if they inherit the mutation. — Figure 399-2A: Typical pheochromocytoma (adrenal unilateral) on magnetic resonance imaging (MRI).
Figure 3¶
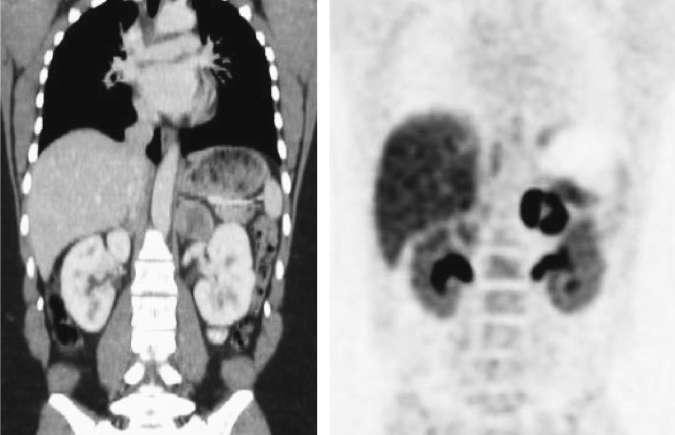
Caption: FIGURE 399-2 Typical pheochromocytoma (adrenal unilateral). A. Magnetic resonance (Part A was provided courtesy of Dr. Tobias Krauss, Freiburg. Part B was provided Differential Diagnosis When the possibility of a pheochromocy- toma is being entertained, other disorders to consider include essential hypertension, anxiety attacks, use of cocaine or amphetamines, mastocy- tosis or carcinoid syndrome (usually without hypertension), intracranial lesions, clonidine withdrawal, autonomic epilepsy, and factitious crises (usually from use of sympathomimetic amines). When an asymptomatic adrenal mass is identified, likely diagnoses other than pheochromocy- of — Figure 399-2B: Typical pheochromocytoma (adrenal unilateral) on 18F-DOPA positron emission tomography (PET).
Figure 4¶

Caption: FIGURE 399-4 Multiple and metastatic pheochromocytoma. A. Paraganglioma emission tomography (PET) demonstrating tumor uptake in the right jugular glomus, the (arrows). Note the physiologic accumulation of the radiopharmaceutical agent in the Several metastases marked by arrows. (Parts A and B were provided courtesy of Dr. Juri — Figure 399-3A: Carotid body tumor (head and neck paraganglioma).
Figure 5¶

Caption: FIGURE 399-4 Multiple and metastatic pheochromocytoma. A. Paraganglioma emission tomography (PET) demonstrating tumor uptake in the right jugular glomus, the (arrows). Note the physiologic accumulation of the radiopharmaceutical agent in the Several metastases marked by arrows. (Parts A and B were provided courtesy of Dr. Juri — Figure 399-3B: Thoracic tumor (extra-adrenal pheochromocytoma).
Figure 6¶
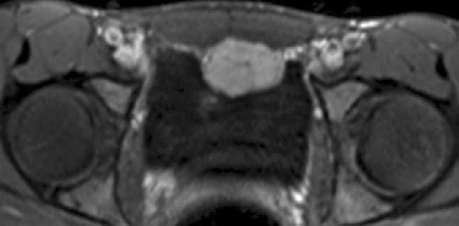
Caption: FIGURE 399-2 Typical pheochromocytoma (adrenal unilateral). A. Magnetic resonance (Part A was provided courtesy of Dr. Tobias Krauss, Freiburg. Part B was provided Differential Diagnosis When the possibility of a pheochromocy- toma is being entertained, other disorders to consider include essential hypertension, anxiety attacks, use of cocaine or amphetamines, mastocy- tosis or carcinoid syndrome (usually without hypertension), intracranial lesions, clonidine withdrawal, autonomic epilepsy, and factitious crises (usually from use of sympathomimetic amines). When an asymptomatic adrenal mass is identified, likely diagnoses other than pheochromocy- of — Figure 399-3C: Paraaortal tumor (extra-adrenal pheochromocytoma).
Figure 7¶
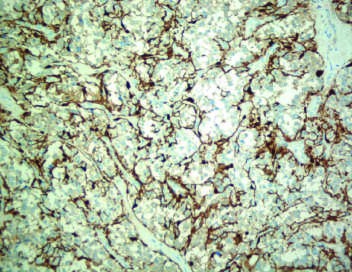
Caption: FIGURE 399-4 Multiple and metastatic pheochromocytoma. A. Paraganglioma emission tomography (PET) demonstrating tumor uptake in the right jugular glomus, the (arrows). Note the physiologic accumulation of the radiopharmaceutical agent in the Several metastases marked by arrows. (Parts A and B were provided courtesy of Dr. Juri — Figure 399-3D: Pelvic tumor at the anterior wall of the urinary bladder (extra-adrenal pheochromocytoma).
Figure 8¶

Caption: FIGURE 399-4 Multiple and metastatic pheochromocytoma. A. Paraganglioma emission tomography (PET) demonstrating tumor uptake in the right jugular glomus, the (arrows). Note the physiologic accumulation of the radiopharmaceutical agent in the Several metastases marked by arrows. (Parts A and B were provided courtesy of Dr. Juri — Figure 399-4A: Paraganglioma syndrome (SDHD W5X mutation) on 68Ga-DOTATATE PET showing tumor uptake in jugular glomus, carotid body, adrenal glands, and interaortocaval paraganglion.
Figure 9¶
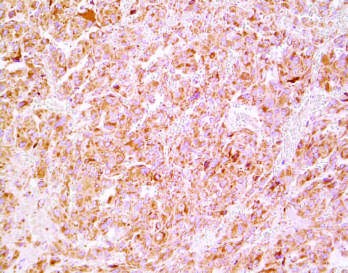
Caption: FIGURE 399-4 Multiple and metastatic pheochromocytoma. A. Paraganglioma emission tomography (PET) demonstrating tumor uptake in the right jugular glomus, the (arrows). Note the physiologic accumulation of the radiopharmaceutical agent in the Several metastases marked by arrows. (Parts A and B were provided courtesy of Dr. Juri — Figure 399-4B: 18F-DOPA PET of a patient with metastatic pheochromocytoma showing several metastases.
Figure 10¶

Caption: FIGURE 399-4 Multiple and metastatic pheochromocytoma. A. Paraganglioma emission tomography (PET) demonstrating tumor uptake in the right jugular glomus, the (arrows). Note the physiologic accumulation of the radiopharmaceutical agent in the Several metastases marked by arrows. (Parts A and B were provided courtesy of Dr. Juri — Figure 399-5A: Hematoxylin and eosin staining of pheochromocytoma histology.
Figure 11¶

Caption: FIGURE 399-4 Multiple and metastatic pheochromocytoma. A. Paraganglioma emission tomography (PET) demonstrating tumor uptake in the right jugular glomus, the (arrows). Note the physiologic accumulation of the radiopharmaceutical agent in the Several metastases marked by arrows. (Parts A and B were provided courtesy of Dr. Juri — Figure 399-5B: Chromogranin staining of pheochromocytoma chief cells.
Figure 12¶

Caption: FIGURE 399-2 Typical pheochromocytoma (adrenal unilateral). A. Magnetic resonance (Part A was provided courtesy of Dr. Tobias Krauss, Freiburg. Part B was provided Differential Diagnosis When the possibility of a pheochromocy- toma is being entertained, other disorders to consider include essential hypertension, anxiety attacks, use of cocaine or amphetamines, mastocy- tosis or carcinoid syndrome (usually without hypertension), intracranial lesions, clonidine withdrawal, autonomic epilepsy, and factitious crises (usually from use of sympathomimetic amines). When an asymptomatic adrenal mass is identified, likely diagnoses other than pheochromocy- of — Figure 399-5C: Synaptophysin staining of pheochromocytoma chief cells.
Figure 13¶

Caption: FIGURE 399-4 Multiple and metastatic pheochromocytoma. A. Paraganglioma emission tomography (PET) demonstrating tumor uptake in the right jugular glomus, the (arrows). Note the physiologic accumulation of the radiopharmaceutical agent in the Several metastases marked by arrows. (Parts A and B were provided courtesy of Dr. Juri — Figure 399-5D: S-100 staining of pheochromocytoma sustentacular cells.
Figure 14¶

Caption: FIGURE 399-2 Typical pheochromocytoma (adrenal unilateral). A. Magnetic resonance (Part A was provided courtesy of Dr. Tobias Krauss, Freiburg. Part B was provided Differential Diagnosis When the possibility of a pheochromocy- toma is being entertained, other disorders to consider include essential hypertension, anxiety attacks, use of cocaine or amphetamines, mastocy- tosis or carcinoid syndrome (usually without hypertension), intracranial lesions, clonidine withdrawal, autonomic epilepsy, and factitious crises (usually from use of sympathomimetic amines). When an asymptomatic adrenal mass is identified, likely diagnoses other than pheochromocy- of — Figure 399-5E/F: Immunohistochemistry with SDHB antibody showing positive (intact) and negative (absent) staining indicating germline mutation status.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.