Chapter 450 | Ataxic Disorders¶
Chapter 450 | Part 13: Neurologic Disorders · Part 13 – Neurologic Disorders
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- Symmetric ataxia is classified by onset: acute (hours/days), subacute (weeks/months), or chronic (months/years).
- Focal ataxia with headache, impaired consciousness, and ipsilateral cranial nerve palsies implies a space-occupying cerebellar lesion.
- Acute reversible ataxias include intoxication (alcohol, phenytoin, lithium, barbiturates, solvents) and postinfectious syndromes.
- Subacute ataxia suggests degeneration of the cerebellar vermis (alcoholism + malnutrition, vitamin B1/B12 deficiency, hyponatremia, paraneoplastic syndromes, prion disorders).
- Chronic symmetric gait ataxia suggests inherited ataxia, metabolic disorder, or chronic infection (e.g., syphilis, Lyme).
- Autosomal dominant spinocerebellar ataxias (SCAs) are caused by CAG triplet repeat expansions resulting in toxic polyglutamine proteins (ataxins).
- Friedreich's ataxia is the most common inherited ataxia, caused by GAA triplet repeats in the frataxin gene leading to mitochondrial iron accumulation and ATP deficiency.
- Ataxia Telangiectasia (AT) is caused by ATM gene mutations, defective DNA repair, and carries high risk for lymphomas, breast cancer, and endocrine disorders.
- Omaveloxolone is the only FDA-approved agent for Friedreich's ataxia, acting as an NRF2 agonist to address mitochondrial dysfunction.
- Prion disease treatment aims to reduce PrPC levels rather than directly eliminating PrPSc, as this method is independent of the pathogenic PrPSc form.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 Classification by Onset
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Autosomal Dominant Ataxias (SCAs)
- 3.2 Autosomal Recessive Ataxias
- 4. CLINICAL FEATURES
- 4.1 Specific SCA Phenotypes
- 5. DIFFERENTIAL DIAGNOSIS
- 5.1 Symmetric vs Focal Ataxia
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Diagnostic Algorithm
- 7. MANAGEMENT & TREATMENT
- 7.1 Pharmacologic Treatment
- 7.2 Genetic Counseling
- 8. PROGNOSIS & COMPLICATIONS
- 8.1 Prognostic Factors
- 9. SPECIAL CONSIDERATIONS
- 10. KEY PEARLS & CLINICAL TRAPS
- WHAT TO LOOK FOR — DIAGNOSTIC CLUES
- WHAT EXCLUDES THE DIAGNOSIS
- TABLES
- Table 450-1: Etiology of Cerebellar Ataxia
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🖼 Figure | Sagittal magnetic resonance imaging (MRI) of the brain of a 60-year- old... |
| 2 | 🖼 Figure | Sagittal magnetic resonance imaging (MRI) of the brain and spinal cord of... |
1. DEFINITION & OVERVIEW¶
- Ataxic disorders are characterized by gait impairment, unclear (scanning) speech, visual blurring due to nystagmus, hand incoordination, and tremor with movement.
- These symptoms result from involvement of the cerebellum and its afferent and efferent pathways, including the spinocerebellar pathways, and the frontopontocerebellar pathway originating in the rostral frontal lobe.
- True cerebellar ataxia must be distinguished from ataxia associated with vestibular nerve or labyrinthine disease, as the latter results in a disorder of gait associated with significant dizziness, light-headedness, or the perception of movement.
- True cerebellar ataxia is devoid of vertiginous complaints and is clearly an unsteady gait due to imbalance.
- Harrison's defines symmetric ataxia as: 'A gradual and progressive increase in symptoms with bilateral and symmetric involvement suggests a genetic, metabolic, immune, or toxic etiology.'
- Harrison's defines focal ataxia as: 'Focal, unilateral symptoms with headache and impaired level of consciousness accompanied by ipsilateral cranial nerve palsies and contralateral weakness imply a space-occupying cerebellar lesion.'
- Symmetric and progressive ataxia can be classified with respect to onset as acute (over hours or days), subacute (weeks or months), or chronic (months to years).
- Acute and reversible ataxias include those caused by intoxication with alcohol, phenytoin, lithium, barbiturates, and other drugs.
- Intoxication caused by toluene exposure, gasoline sniffing, glue sniffing, spray painting, or exposure to methyl mercury or bismuth are additional causes of acute or subacute ataxia.
- Treatment with cytotoxic chemotherapeutic drugs such as fluorouracil and paclitaxel can cause acute or subacute ataxia.
- Patients with a postinfectious syndrome (especially after varicella) may develop gait ataxia and mild dysarthria, both of which are reversible.
- Rare infectious causes of acquired ataxia include poliovirus, coxsackievirus, echovirus, Epstein-Barr virus, toxoplasmosis, Legionella, and Lyme disease.
- The subacute development of ataxia of gait over weeks to months (degeneration of the cerebellar vermis) may be due to the combined effects of alcoholism and malnutrition, particularly with deficiencies of vitamins B1 and B12.
- Hyponatremia has also been associated with ataxia.
- Paraneoplastic cerebellar ataxia is associated with a number of different tumors (and autoantibodies) such as breast and ovarian cancers (anti-Yo), small-cell lung cancer (anti-PQ-type voltage-gated calcium channel), and Hodgkin's disease (anti-Tr).
- Another paraneoplastic syndrome associated with myoclonus and opsoclonus occurs with breast (anti-Ri) and lung cancers and neuroblastoma.
- For all of these paraneoplastic ataxias, the neurologic syndrome may be the presenting symptom of the cancer.
- Autoantibody-associated cerebellar syndromes also occur without a cancer association. The most common is a progressive ataxic syndrome affecting speech and gait associated with serum anti-glutamic acid decarboxylase (GAD65) antibodies.
- Another immune-mediated progressive ataxia is associated with antigliadin (and antiendomysium) antibodies and the human leukocyte antigen (HLA) DQB1*0201 haplotype; in some affected patients, biopsy of the small intestine reveals villus atrophy consistent with gluten-sensitive enteropathy.
- Subacute progressive ataxia may be caused by a prion disorder, especially when an infectious etiology, such as transmission from contaminated human growth hormone, is responsible.
- Chronic symmetric gait ataxia suggests an inherited ataxia (discussed below), a metabolic disorder, or a chronic infection.
- Hypothyroidism must always be considered as a readily treatable and reversible form of gait ataxia.
- Infectious diseases that can present with ataxia are meningovascular syphilis and tabes dorsalis due to degeneration of the posterior columns and spinocerebellar pathways in the spinal cord.
1.1 Classification by Onset¶
- Acute (Hours to Days): Intoxication (alcohol, lithium, phenytoin, barbiturates), Acute viral cerebellitis, Postinfection syndrome.
- Subacute (Days to Weeks): Intoxication (mercury, solvents, gasoline, glue, cytotoxic chemotherapeutic drugs), Alcoholic-nutritional (vitamin B1 and B12 deficiency), Lyme disease, Phenytoin toxicity, Amiodarone.
- Chronic (Months to Years): Inherited diseases, Tabes dorsalis (tertiary syphilis), Hypothyroidism, AIDS-related multifocal leukoencephalopathy, Stable gliosis secondary to vascular lesion or demyelinating plaque, Congenital lesion (Chiari or Dandy-Walker malformations).
- Focal and Ipsilateral Cerebellar Signs: Vascular (cerebellar infarction, hemorrhage, subdural hematoma), Neoplastic (cerebellar glioma or metastatic tumor), Infectious (cerebellar abscess), Demyelinating (multiple sclerosis), AIDS-related multifocal leukoencephalopathy, Congenital lesion (Chiari or Dandy-Walker malformations).
2. EPIDEMIOLOGY¶
- Ataxias with autosomal dominant, autosomal recessive, X-linked, or mitochondrial forms of inheritance are present on a worldwide basis.
- Machado-Joseph disease (SCA3) (autosomal dominant) and Friedreich's ataxia (autosomal recessive) are the most common types in most populations.
- Friedreich's ataxia composes one-half of all hereditary ataxias.
- Genetic markers are now commercially available to precisely identify the genetic mutation for correct diagnosis and also for family planning.
- Early detection of asymptomatic preclinical disease can reduce or eliminate the inherited form of ataxia in some families on a global, worldwide basis.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- Prion Diseases: The best method to treat human prion diseases may be by reducing PrPC, the substrate for PrPSc, rather than trying to directly reduce or eliminate PrPSc.
- The lifespan of mice is normal when PrPC is reduced or even eliminated; when PRNP is knocked out, mice live a normal span of time with minimal deficits detected, aside from a mild neuropathy.
- Identification of adults hemizygous for PRNP suggests that humans can also live with reduced levels of PrPC.
- Currently, antisense oligonucleotides (ASOs) against PrPC are being tested in symptomatic prion disease.
- One advantage of treatment methods reducing PrPC is that if they are effective, they should work for all strains and types of human prion disease, as the method is independent of the form of PrPSc that is pathogenic in a given individual.
- Autosomal Dominant Ataxias: The autosomal spinocerebellar ataxias (SCAs) include SCA types 1 through 50, dentatorubropallidoluysian atrophy (DRPLA), and episodic ataxia (EA) types 1 to 7.
- SCA1, SCA2, SCA3 (Machado-Joseph disease [MJD]), SCA6, SCA7, and SCA17 are caused by CAG triplet repeat expansions in different genes.
- SCA8 is due to an untranslated CTG repeat expansion, SCA12 is linked to an untranslated CAG repeat, and SCA10 is caused by an untranslated pentanucleotide repeat.
- The clinical phenotypes of these SCAs overlap.
- The genotype has become the gold standard for diagnosis and classification.
- CAG encodes glutamine, and these expanded CAG triplet repeat expansions result in expanded polyglutamine proteins, termed ataxins, that produce a toxic gain of function with autosomal dominant inheritance.
- Although the phenotype is variable for any given disease gene, a pattern of neuronal loss with gliosis is produced that is relatively unique for each ataxia.
- Mechanism of Polyglutamine Ataxins: Expanded polyglutamine ataxins with more than ~40 glutamines are potentially toxic to neurons for a variety of reasons including the following:
- High levels of gene expression for the mutant polyglutamine ataxin in affected neurons.
- Conformational change of the aggregated protein to a β-pleated structure.
- Abnormal transport of the ataxin into the nucleus (SCA1, MJD, SCA7).
- Binding to other polyglutamine proteins, including the TATA-binding transcription protein and the CREB-binding protein, impairing their functions.
- Impairing the efficiency of the ubiquitin-proteasome system of protein turnover.
- Inducing neuronal apoptosis.
- An earlier age of onset (anticipation) and more aggressive disease in subsequent generations are due to further expansion of the CAG triplet repeat and increased polyglutamine number in the mutant ataxin.
- Friedreich's Ataxia: This is the most common form of inherited ataxia.
- The classic form of Friedreich's ataxia has been mapped to 9q13-q21.1, and the mutant gene, frataxin, contains expanded GAA triplet repeats in the first intron.
- There is homozygosity for expanded GAA repeats in >95% of patients.
- Normal persons have 7-22 GAA repeats, and patients have 200-900 GAA repeats.
- A more varied clinical syndrome has been described in compound heterozygotes who have one copy of the GAA expansion and the other copy a point mutation in the frataxin gene.
- When the point mutation is located in the region of the gene that encodes the amino-terminal half of frataxin, the phenotype is milder, often consisting of a spastic gait, retained or exaggerated reflexes, no dysarthria, and mild or absent ataxia.
- Patients with Friedreich's ataxia have undetectable or extremely low levels of frataxin mRNA, as compared with carriers and unrelated individuals; thus, disease appears to be caused by a loss of expression of the frataxin protein.
- Frataxin is a mitochondrial protein involved in iron homeostasis.
- Mitochondrial iron accumulation due to loss of the iron transporter coded by the mutant frataxin gene results in a deficiency in iron/sulfur clusters containing mitochondrial enzymes, decreased ATP production, and accumulation of iron in the heart.
- Excess oxidized iron results in turn in the oxidation of cellular components and irreversible cell injury.
- Vitamin E Deficiency Syndromes: Two forms of hereditary ataxia associated with abnormalities in the interactions of vitamin E (α-tocopherol) with very-low-density lipoprotein (VLDL) have been delineated.
- These are abetalipoproteinemia (Bassen-Kornzweig syndrome) and ataxia with vitamin E deficiency (AVED).
- Abetalipoproteinemia is caused by mutations in the gene coding for the larger subunit of the microsomal triglyceride transfer protein (MTP).
- Defects in MTP result in impairment of formation and secretion of VLDL in liver.
- This defect results in a deficiency of vitamin E delivery to tissues, including the central and peripheral nervous system, as VLDL is the transport molecule for vitamin E and other fat-soluble substitutes.
- AVED is due to mutations in the gene for α-tocopherol transfer protein (α-TTP).
- These patients have an impaired ability to bind vitamin E into the VLDL produced and secreted by the liver, resulting in a deficiency of vitamin E in peripheral tissues.
- Ataxia Telangiectasia: The gene for AT (the ATM gene) at 11q22-23 encodes a protein that is similar to several yeast and mammalian phosphatidylinositol-3' kinases involved in mitogenic signal transduction, meiotic recombination, and cell cycle control.
- Defective DNA repair in AT fibroblasts exposed to ultraviolet light has been demonstrated.
- The discovery of ATM permits early diagnosis and identification of heterozygotes who are at risk for cancer (e.g., breast cancer).
- Mitochondrial Ataxias: Spinocerebellar syndromes have been identified with mutations in mitochondrial DNA (mtDNA).
- Thirty pathogenic mtDNA point mutations and 60 different types of mtDNA deletions are known, several of which cause or are associated with ataxia.
- Episodic Ataxia: EA types 1 and 2 are two rare dominantly inherited disorders that have been mapped to chromosomes 12p (a potassium channel gene, KCNA1, Phe249Leu mutation) for type 1 and 19p for type 2.
- Patients with EA-1 have brief episodes of ataxia with myokymia and nystagmus that last only minutes. Startle, sudden change in posture, and exercise can induce episodes.
- Patients with EA-2 have episodes of ataxia with nystagmus that can last for hours or days. Stress, exercise, or excessive fatigue may be precipitants.
- Stop codon, nonsense mutations causing EA-2 have been found in the CACNA1A gene, encoding the α voltage-dependent 1A calcium channel subunit.
3.1 Autosomal Dominant Ataxias (SCAs)¶
- SCA1: Previously referred to as olivopontocerebellar atrophy, but genomic data have shown that that entity represents several different genotypes with overlapping clinical features.
- SCA1 encodes a gene product, called ataxin-1, that regulates transcriptional repression with various nuclear factors.
- As a protein that can bind RNA, ataxin-1 may also regulate gene transcription posttranslationally.
- The mutant allele has 40 CAG repeats located within the coding region, whereas alleles from unaffected individuals have ≤36 repeats.
- A few patients with 38-40 CAG repeats have been described.
- There is a direct correlation between a larger number of repeats and a younger age of onset for SCA1.
- Juvenile patients have higher numbers of repeats, and anticipation is present in subsequent generations.
- SCA2: The gene for MJD maps to 14q24.3-q32.
- Unstable CAG repeat expansions are present in the MJD gene coding for a polyglutamine-containing protein named ataxin-3, or MJD-ataxin.
- An earlier age of onset is associated with longer repeats.
- Alleles from normal individuals have between 12 and 37 CAG repeats, whereas MJD alleles have 60-84 CAG repeats.
- Polyglutamine-containing aggregates of ataxin-3 (MJD-ataxin) have been described in neuronal nuclei undergoing degeneration.
- MJD-ataxin codes for a ubiquitin protease, which is inactive due to expanded polyglutamines.
- Proteosome function is impaired, resulting in altered clearance of proteins and cerebellar neuronal loss.
- SCA3 (Machado-Joseph Disease): MJD was first described among the Portuguese and their descendants in New England and California.
- Subsequently, MJD has been found in families from Portugal, Australia, Brazil, Canada, China, England, France, India, Israel, Italy, Japan, Spain, Taiwan, and the United States.
- In most populations, it is the most common autosomal dominant ataxia.
- MJD has been classified into three clinical types.
- Type I MJD (ALS-parkinsonism-dystonia type): Neurologic deficits appear in the first two decades and involve weakness and spasticity of extremities, especially the legs, often with dystonia of the face, neck, trunk, and extremities.
- Patellar and ankle clonus are common, as are extensor plantar responses.
- The gait is slow and stiff, with a slightly broadened base and lurching from side to side; this gait results from spasticity, not true ataxia.
- There is no truncal titubation.
- Pharyngeal weakness and spasticity cause difficulty with speech and swallowing.
- Of note is the prominence of horizontal and vertical nystagmus, loss of fast saccadic eye movements, hypermetric and hypometric saccades, and impairment of upward vertical gaze.
- Facial fasciculations, facial myokymia, lingual fasciculations without atrophy, ophthalmoparesis, and ocular prominence are common early manifestations.
- Type II MJD (ataxic type): True cerebellar deficits of dysarthria and gait and extremity ataxia begin in the second to fourth decades along with corticospinal and extrapyramidal deficits of spasticity, rigidity, and dystonia.
- Type II is the most common form of MJD.
- Ophthalmoparesis, upward vertical gaze deficits, and facial and lingual fasciculations are also present.
- Type III MJD (ataxic-amyotrophic type): Presents in the fifth to seventh decades with a pancerebellar disorder that includes dysarthria and gait and extremity ataxia.
- Distal sensory loss involving pain, touch, vibration, and position senses and distal atrophy are prominent, indicating the presence of peripheral neuropathy.
- The deep tendon reflexes are depressed to absent, and there are no corticospinal or extrapyramidal findings.
- The mean age of onset of symptoms in MJD is 25 years.
- Neurologic deficits invariably progress and lead to death from debilitation within 15 years of onset, especially in patients with types I and II disease.
- Usually, patients retain full intellectual function.
- SCA6: CAG repeat expansions (21-27 in patients; 4-16 triplets in normal individuals) result in late-onset progressive ataxia with cerebellar degeneration.
- Missense mutations in this gene result in familial hemiplegic migraine.
- Nonsense mutations resulting in termination of protein synthesis of the gene product yield hereditary paroxysmal cerebellar ataxia or EA.
- Some patients with familial hemiplegic migraine develop progressive ataxia and also have cerebellar atrophy.
- SCA7: This disorder is distinguished from all other SCAs by the presence of retinal pigmentary degeneration.
- The visual abnormalities first appear as blue-yellow color blindness and proceed to frank visual loss with macular degeneration.
- In almost all other respects, SCA7 resembles several other SCAs in which ataxia is accompanied by various non-cerebellar findings, including ophthalmoparesis and extensor plantar responses.
- The genetic defect is an expanded CAG repeat in the SCA7 gene at 3p14-p21.1.
- The expanded repeat size in SCA7 is highly variable.
- Consistent with this, the severity of clinical findings varies from essentially asymptomatic to mild late-onset symptoms to severe, aggressive disease in childhood with rapid progression.
- Marked anticipation has been recorded, especially with paternal transmission.
- The disease protein, ataxin-7, forms aggregates in nuclei of affected neurons, as has also been described for SCA1 and SCA3/MJD.
- Ataxin-7 is a subunit of GCN5, a histone acetyltransferase-containing complex.
- SCA8: This form of ataxia is caused by a CTG repeat expansion in an untranslated region of a gene on chromosome 13q21.
- There is marked maternal bias in transmission, perhaps reflecting contractions of the repeat during spermatogenesis.
- The mutation is not fully penetrant.
- Symptoms include slowly progressive dysarthria and gait ataxia beginning at ~40 years of age with a range between 20 and 65 years.
- Other features include nystagmus, leg spasticity, and reduced vibratory sensation.
- Severely affected individuals are nonambulatory by the fourth to sixth decades.
- MRI shows cerebellar atrophy.
- The mechanism of disease may involve a dominant 'toxic' effect occurring at the RNA level, as occurs in myotonic dystrophy.
- SCA27B: SCA27B is a recently discovered entity, resulting from an intronic GAA repeat expansion in the FGF14 gene, and is one of the most common late-onset inherited ataxias.
- SCA27B occurs with a median age of onset of 60 years and presents as a relatively pure cerebellar ataxia with episodic symptoms at the disease onset.
- Other clinical features include afferent sensory deficits and dysautonomia.
- Cognitive impairment is infrequent.
- Both the episodic symptoms and symptom severity of ataxia appear to improve with 4-aminopyridine, although randomized clinical trials have not yet been performed in this population.
- DRPLA: DRPLA has a variable presentation that may include progressive ataxia, choreoathetosis, dystonia, seizures, myoclonus, and dementia.
- DRPLA is due to unstable CAG triplet repeats in the open reading frame of a gene named atrophin located on chromosome 12p12-ter.
- Larger expansions are found in patients with earlier onset.
- The number of repeats is 49 in patients with DRPLA and ≤26 in normal individuals.
- Anticipation occurs in successive generations, with earlier onset of disease in association with an increasing CAG repeat number in children who inherit the disease from their father.
- One well-characterized family in North Carolina has a phenotypic variant known as the Haw River syndrome, now recognized to be due to the DRPLA mutation.
3.2 Autosomal Recessive Ataxias¶
- Friedreich's Ataxia: This is the most common form of inherited ataxia, composing one-half of all hereditary ataxias.
- It can occur in a classic form or in association with a genetically determined vitamin E deficiency syndrome; the two forms are clinically indistinguishable.
- The classic form of Friedreich's ataxia has been mapped to 9q13-q21.1, and the mutant gene, frataxin, contains expanded GAA triplet repeats in the first intron.
- There is homozygosity for expanded GAA repeats in >95% of patients.
- Normal persons have 7-22 GAA repeats, and patients have 200-900 GAA repeats.
- A more varied clinical syndrome has been described in compound heterozygotes who have one copy of the GAA expansion and the other copy a point mutation in the frataxin gene.
- When the point mutation is located in the region of the gene that encodes the amino-terminal half of frataxin, the phenotype is milder, often consisting of a spastic gait, retained or exaggerated reflexes, no dysarthria, and mild or absent ataxia.
- Patients with Friedreich's ataxia have undetectable or extremely low levels of frataxin mRNA, as compared with carriers and unrelated individuals; thus, disease appears to be caused by a loss of expression of the frataxin protein.
- Frataxin is a mitochondrial protein involved in iron homeostasis.
- Mitochondrial iron accumulation due to loss of the iron transporter coded by the mutant frataxin gene results in a deficiency in iron/sulfur clusters containing mitochondrial enzymes, decreased ATP production, and accumulation of iron in the heart.
- Excess oxidized iron results in turn in the oxidation of cellular components and irreversible cell injury.
- Vitamin E Deficiency Syndromes: Two forms of hereditary ataxia associated with abnormalities in the interactions of vitamin E (α-tocopherol) with very-low-density lipoprotein (VLDL) have been delineated.
- These are abetalipoproteinemia (Bassen-Kornzweig syndrome) and ataxia with vitamin E deficiency (AVED).
- Abetalipoproteinemia is caused by mutations in the gene coding for the larger subunit of the microsomal triglyceride transfer protein (MTP).
- Defects in MTP result in impairment of formation and secretion of VLDL in liver.
- This defect results in a deficiency of vitamin E delivery to tissues, including the central and peripheral nervous system, as VLDL is the transport molecule for vitamin E and other fat-soluble substitutes.
- AVED is due to mutations in the gene for α-tocopherol transfer protein (α-TTP).
- These patients have an impaired ability to bind vitamin E into the VLDL produced and secreted by the liver, resulting in a deficiency of vitamin E in peripheral tissues.
- Hence, either absence of VLDL (abetalipoproteinemia) or impaired binding of vitamin E to VLDL (AVED) causes an ataxic syndrome.
- RFC1-Related CANVAS Syndrome: Biallelic intronic AAGGG repeat expansions in the replication factor C subunit 1 (RFC1) gene are the cause of late-onset ataxia, particularly if associated with sensory neuronopathy and bilateral vestibular areflexia (CANVAS syndrome).
- A chronic unexplained cough is often associated with and may precede the onset of neurologic symptoms.
- Ataxia Telangiectasia: Patients with ataxia telangiectasia (AT) present in the first decade of life with progressive telangiectatic lesions associated with deficits in cerebellar function and nystagmus.
- The neurologic manifestations correspond to those in Friedreich's disease, which should be included in the differential diagnosis.
- Truncal and limb ataxia, dysarthria, extensor plantar responses, myoclonic jerks, areflexia, and distal sensory deficits may develop.
- There is a high incidence of recurrent pulmonary infections and neoplasms of the lymphatic and reticuloendothelial system in patients with AT.
- Thymic hypoplasia with cellular and humoral (IgA and IgG2) immunodeficiencies, premature aging, and endocrine disorders such as type 1 diabetes mellitus are described.
- There is an increased incidence of lymphomas, Hodgkin's disease, acute T-cell leukemias, and breast cancer.
- The most striking neuropathologic changes include loss of Purkinje, granule, and basket cells in the cerebellar cortex as well as of neurons in the deep cerebellar nuclei.
- The inferior olives of the medulla may also have neuronal loss.
- There is a loss of anterior horn neurons in the spinal cord and of dorsal root ganglion cells associated with posterior column spinal cord demyelination.
- A poorly developed or absent thymus gland is the most consistent defect of the lymphoid system.
- Elevated serum alpha-fetoprotein and immunoglobulin deficiency are noted.
- Mitochondrial Ataxias: Spinocerebellar syndromes have been identified with mutations in mitochondrial DNA (mtDNA).
- Thirty pathogenic mtDNA point mutations and 60 different types of mtDNA deletions are known, several of which cause or are associated with ataxia.
4. CLINICAL FEATURES¶
- Symptoms and signs of ataxia consist of gait impairment, unclear (scanning) speech, visual blurring due to nystagmus, hand incoordination, and tremor with movement.
- These result from the involvement of the cerebellum and its afferent and efferent pathways, including the spinocerebellar pathways, and the frontopontocerebellar pathway originating in the rostral frontal lobe.
- True cerebellar ataxia must be distinguished from ataxia associated with vestibular nerve or labyrinthine disease, as the latter results in a disorder of gait associated with a significant degree of dizziness, light-headedness, or the perception of movement.
- True cerebellar ataxia is devoid of these vertiginous complaints and is clearly an unsteady gait due to imbalance.
- Symmetric Ataxia: A gradual and progressive increase in symptoms with bilateral and symmetric involvement suggests a genetic, metabolic, immune, or toxic etiology.
- Focal Ataxia: Focal, unilateral symptoms with headache and impaired level of consciousness accompanied by ipsilateral cranial nerve palsies and contralateral weakness imply a space-occupying cerebellar lesion.
- Acute Focal Ataxia: Commonly results from cerebrovascular disease, usually ischemic infarction or cerebellar hemorrhage.
- These lesions typically produce cerebellar symptoms ipsilateral to the injured cerebellum and may be associated with an impaired level of consciousness due to brainstem compression and increased intracranial pressure.
- Ipsilateral pontine signs, including sixth and seventh nerve palsies, may be present.
- Focal and worsening signs of acute ataxia should also prompt consideration of a posterior fossa subdural hematoma, bacterial abscess, or primary or metastatic cerebellar tumor.
- Chronic Etiologies: Chronic etiologies of progressive ataxia include multiple sclerosis and congenital lesions such as a Chiari malformation or a congenital cyst of the posterior fossa (Dandy-Walker syndrome).
- Inherited Ataxias: Inherited ataxias may show autosomal dominant, autosomal recessive, or maternal (mitochondrial) modes of inheritance.
- Although the clinical manifestations and neuropathologic findings of cerebellar disease dominate the clinical picture, there may also be characteristic changes in the basal ganglia, brainstem, spinal cord, optic nerves, retina, and peripheral nerves.
- In large families with dominantly inherited ataxias, many gradations are observed from purely cerebellar manifestations to mixed cerebellar and brainstem disorders, cerebellar and basal ganglia syndromes, and spinal cord or peripheral nerve disease.
- Rarely, dementia is present as well.
- The clinical picture may be homogeneous within a family with dominantly inherited ataxia, but sometimes most affected family members show one characteristic syndrome, while one or several members have an entirely different phenotype.
- SCA1 Symptoms and Signs: SCA1 is characterized by the development in early- or middle-adult life of progressive cerebellar ataxia of the trunk and limbs, impairment of equilibrium and gait, slowness of voluntary movements, scanning speech, nystagmoid eye movements, and oscillatory tremor of the head and trunk.
- Dysarthria, dysphagia, and oculomotor and facial palsies may also occur.
- Extrapyramidal symptoms include rigidity, an immobile face, and parkinsonian tremor.
- The reflexes are usually normal, but knee and ankle jerks may be lost, and extensor plantar responses may occur.
- Dementia may be noted but is usually mild.
- Impairment of sphincter function is common, with urinary and sometimes fecal incontinence.
- Cerebellar and brainstem atrophy are evident on MRI.
- Marked shrinkage of the ventral half of the pons, disappearance of the olivary eminence on the ventral surface of the medulla, and atrophy of the cerebellum are evident on gross postmortem inspection of the brain.
- Variable loss of Purkinje cells, reduced numbers of cells in the molecular and granular layer, demyelination of the middle cerebellar peduncle and the cerebellar hemispheres, and severe loss of cells in the pontine nuclei and olives are found on histologic examination.
- Degenerative changes in the striatum, especially the putamen, and loss of the pigmented cells of the substantia nigra may be found in cases with extrapyramidal features.
- More widespread degeneration in the central nervous system (CNS), including involvement of the posterior columns and the spinocerebellar fibers, is often present.
- SCA2 Symptoms and Signs: Another clinical phenotype, SCA2, has been described in patients from Cuba and India.
- Cuban patients probably are descendants of a common ancestor, and the population may be the largest homogeneous group of patients with ataxia described.
- The age of onset ranges from 2 to 65 years, and there is considerable clinical variability within families.
- Although neuropathologic and clinical findings are compatible with a diagnosis of SCA1, including slow saccadic eye movements, ataxia, dysarthria, parkinsonian rigidity, optic disc pallor, mild spasticity, and retinal degeneration, SCA2 is a unique form of cerebellar degenerative disease.
- Type II MJD (ataxic type): True cerebellar deficits of dysarthria and gait and extremity ataxia begin in the second to fourth decades along with corticospinal and extrapyramidal deficits of spasticity, rigidity, and dystonia.
- Type II is the most common form of MJD.
- Ophthalmoparesis, upward vertical gaze deficits, and facial and lingual fasciculations are also present.
- Type III MJD (ataxic-amyotrophic type): Presents in the fifth to seventh decades with a pancerebellar disorder that includes dysarthria and gait and extremity ataxia.
- Distal sensory loss involving pain, touch, vibration, and position senses and distal atrophy are prominent, indicating the presence of peripheral neuropathy.
- The deep tendon reflexes are depressed to absent, and there are no corticospinal or extrapyramidal findings.
- The mean age of onset of symptoms in MJD is 25 years.
- Neurologic deficits invariably progress and lead to death from debilitation within 15 years of onset, especially in patients with types I and II disease.
- Usually, patients retain full intellectual function.
- SCA7 Symptoms and Signs: This disorder is distinguished from all other SCAs by the presence of retinal pigmentary degeneration.
- The visual abnormalities first appear as blue-yellow color blindness and proceed to frank visual loss with macular degeneration.
- In almost all other respects, SCA7 resembles several other SCAs in which ataxia is accompanied by various non-cerebellar findings, including ophthalmoparesis and extensor plantar responses.
- The genetic defect is an expanded CAG repeat in the SCA7 gene at 3p14-p21.1.
- The expanded repeat size in SCA7 is highly variable.
- Consistent with this, the severity of clinical findings varies from essentially asymptomatic to mild late-onset symptoms to severe, aggressive disease in childhood with rapid progression.
- Marked anticipation has been recorded, especially with paternal transmission.
- The disease protein, ataxin-7, forms aggregates in nuclei of affected neurons, as has also been described for SCA1 and SCA3/MJD.
- Ataxin-7 is a subunit of GCN5, a histone acetyltransferase-containing complex.
- SCA8 Symptoms: This form of ataxia is caused by a CTG repeat expansion in an untranslated region of a gene on chromosome 13q21.
- There is marked maternal bias in transmission, perhaps reflecting contractions of the repeat during spermatogenesis.
- The mutation is not fully penetrant.
- Symptoms include slowly progressive dysarthria and gait ataxia beginning at ~40 years of age with a range between 20 and 65 years.
- Other features include nystagmus, leg spasticity, and reduced vibratory sensation.
- Severely affected individuals are nonambulatory by the fourth to sixth decades.
- MRI shows cerebellar atrophy.
- The mechanism of disease may involve a dominant 'toxic' effect occurring at the RNA level, as occurs in myotonic dystrophy.
- SCA27B Symptoms: SCA27B is a recently discovered entity, resulting from an intronic GAA repeat expansion in the FGF14 gene, and is one of the most common late-onset inherited ataxias.
- SCA27B occurs with a median age of onset of 60 years and presents as a relatively pure cerebellar ataxia with episodic symptoms at the disease onset.
- Other clinical features include afferent sensory deficits and dysautonomia.
- Cognitive impairment is infrequent.
- Both the episodic symptoms and symptom severity of ataxia appear to improve with 4-aminopyridine, although randomized clinical trials have not yet been performed in this population.
- DRPLA Symptoms: DRPLA has a variable presentation that may include progressive ataxia, choreoathetosis, dystonia, seizures, myoclonus, and dementia.
- DRPLA is due to unstable CAG triplet repeats in the open reading frame of a gene named atrophin located on chromosome 12p12-ter.
- Larger expansions are found in patients with earlier onset.
- The number of repeats is 49 in patients with DRPLA and ≤26 in normal individuals.
- Anticipation occurs in successive generations, with earlier onset of disease in association with an increasing CAG repeat number in children who inherit the disease from their father.
- One well-characterized family in North Carolina has a phenotypic variant known as the Haw River syndrome, now recognized to be due to the DRPLA mutation.
- Episodic Ataxia: Patients with EA-1 have brief episodes of ataxia with myokymia and nystagmus that last only minutes.
- Startle, sudden change in posture, and exercise can induce episodes.
- Acetazolamide or anticonvulsants may be therapeutic.
- Patients with EA-2 have episodes of ataxia with nystagmus that can last for hours or days.
- Stress, exercise, or excessive fatigue may be precipitants.
- Acetazolamide may be therapeutic and can reverse the relative intracellular alkalosis detected by magnetic resonance spectroscopy.
- Stop codon, nonsense mutations causing EA-2 have been found in the CACNA1A gene, encoding the α voltage-dependent 1A calcium channel subunit.
- Friedreich's Ataxia Symptoms and Signs: Friedreich's ataxia presents before 25 years of age with progressive staggering gait, frequent falling, and titubation.
- The lower extremities are more severely involved than the upper ones.
- Ataxia Telangiectasia Symptoms: Patients with ataxia telangiectasia (AT) present in the first decade of life with progressive telangiectatic lesions associated with deficits in cerebellar function and nystagmus.
- The neurologic manifestations correspond to those in Friedreich's disease, which should be included in the differential diagnosis.
- Truncal and limb ataxia, dysarthria, extensor plantar responses, myoclonic jerks, areflexia, and distal sensory deficits may develop.
- There is a high incidence of recurrent pulmonary infections and neoplasms of the lymphatic and reticuloendothelial system in patients with AT.
- Thymic hypoplasia with cellular and humoral (IgA and IgG2) immunodeficiencies, premature aging, and endocrine disorders such as type 1 diabetes mellitus are described.
- There is an increased incidence of lymphomas, Hodgkin's disease, acute T-cell leukemias, and breast cancer.
- Mitochondrial Ataxias: Spinocerebellar syndromes have been identified with mutations in mitochondrial DNA (mtDNA).
- Thirty pathogenic mtDNA point mutations and 60 different types of mtDNA deletions are known, several of which cause or are associated with ataxia.
4.1 Specific SCA Phenotypes¶
- SCA1: Progressive cerebellar ataxia of the trunk and limbs, impairment of equilibrium and gait, slowness of voluntary movements, scanning speech, nystagmoid eye movements, and oscillatory tremor of the head and trunk.
- Dysarthria, dysphagia, and oculomotor and facial palsies may also occur.
- Extrapyramidal symptoms include rigidity, an immobile face, and parkinsonian tremor.
- The reflexes are usually normal, but knee and ankle jerks may be lost, and extensor plantar responses may occur.
- Dementia may be noted but is usually mild.
- Impairment of sphincter function is common, with urinary and sometimes fecal incontinence.
- SCA2: Slow saccadic eye movements, ataxia, dysarthria, parkinsonian rigidity, optic disc pallor, mild spasticity, and retinal degeneration.
- SCA3 (Machado-Joseph Disease): Type I (ALS-parkinsonism-dystonia), Type II (ataxic), Type III (ataxic-amyotrophic).
- SCA7: Distinguished by retinal pigmentary degeneration (blue-yellow color blindness, macular degeneration).
- SCA8: Slowly progressive dysarthria and gait ataxia beginning at ~40 years of age.
- SCA27B: Median age of onset of 60 years, relatively pure cerebellar ataxia with episodic symptoms.
- DRPLA: Progressive ataxia, choreoathetosis, dystonia, seizures, myoclonus, and dementia.
- Episodic Ataxia: EA-1 (minutes, myokymia, nystagmus), EA-2 (hours/days, nystagmus).
- Friedreich's Ataxia: Progressive staggering gait, frequent falling, and titubation before 25 years of age.
- Ataxia Telangiectasia: Progressive telangiectatic lesions, cerebellar function deficits, nystagmus, recurrent pulmonary infections, neoplasms.
5. DIFFERENTIAL DIAGNOSIS¶
- Symmetric Ataxia: A gradual and progressive increase in symptoms with bilateral and symmetric involvement suggests a genetic, metabolic, immune, or toxic etiology.
- Focal Ataxia: Focal, unilateral symptoms with headache and impaired level of consciousness accompanied by ipsilateral cranial nerve palsies and contralateral weakness imply a space-occupying cerebellar lesion.
- Acute Ataxia: Intoxication (alcohol, lithium, phenytoin, barbiturates), Acute viral cerebellitis, Postinfection syndrome.
- Subacute Ataxia: Intoxication (mercury, solvents, gasoline, glue, cytotoxic chemotherapeutic drugs), Alcoholic-nutritional (vitamin B1 and B12 deficiency), Lyme disease, Phenytoin toxicity, Amiodarone.
- Chronic Ataxia: Inherited diseases, Tabes dorsalis (tertiary syphilis), Hypothyroidism, AIDS-related multifocal leukoencephalopathy, Stable gliosis secondary to vascular lesion or demyelinating plaque, Congenital lesion (Chiari or Dandy-Walker malformations).
- Focal and Ipsilateral Cerebellar Signs: Vascular (cerebellar infarction, hemorrhage, subdural hematoma), Neoplastic (cerebellar glioma or metastatic tumor), Infectious (cerebellar abscess), Demyelinating (multiple sclerosis), AIDS-related multifocal leukoencephalopathy, Congenital lesion (Chiari or Dandy-Walker malformations).
- Paraneoplastic Syndromes: Breast and ovarian cancers (anti-Yo), Small-cell lung cancer (anti-PQ-type voltage-gated calcium channel), Hodgkin's disease (anti-Tr), Breast and lung cancers and neuroblastoma (anti-Ri).
- Autoimmune: Anti-glutamic acid decarboxylase (GAD65) antibodies, Antigliadin (and antiendomysium) antibodies with HLA DQB1*0201 haplotype.
- Metabolic: Hypothyroidism, Vitamin E deficiency (AVED, Abetalipoproteinemia), Hyponatremia.
- Infectious: Meningovascular syphilis, Tabes dorsalis, Poliovirus, Coxsackievirus, Echovirus, Epstein-Barr virus, Toxoplasmosis, Legionella, Lyme disease.
- Prion Disorders: Subacute progressive ataxia may be caused by a prion disorder, especially when an infectious etiology, such as transmission from contaminated human growth hormone, is responsible.
5.1 Symmetric vs Focal Ataxia¶
- Symmetric Ataxia: Bilateral and symmetric involvement suggests a genetic, metabolic, immune, or toxic etiology.
- Focal Ataxia: Unilateral symptoms with headache and impaired level of consciousness accompanied by ipsilateral cranial nerve palsies and contralateral weakness imply a space-occupying cerebellar lesion.
- Acute vs Chronic: Acute (hours/days) suggests intoxication or vascular disease. Chronic (months/years) suggests inherited or metabolic disease.
6. INVESTIGATIONS & DIAGNOSIS¶
- Computed tomography (CT) or magnetic resonance imaging (MRI) studies will reveal clinically significant processes of this type.
- Many of these lesions represent true neurologic emergencies, as sudden herniation, either rostrally through the tentorium or caudal herniation of cerebellar tonsils through the foramen magnum, can occur and is usually devastating.
- Acute surgical decompression may be required.
- Lymphoma or progressive multifocal leukoencephalopathy (PML) in a patient with AIDS may present with an acute or subacute focal cerebellar syndrome.
- Chronic etiologies of progressive ataxia include multiple sclerosis and congenital lesions such as a Chiari malformation or a congenital cyst of the posterior fossa (Dandy-Walker syndrome).
- Genetic Diagnostic Laboratories: 1. Baylor College of Medicine; Houston, Texas, 1-713-798-6522 http://www.bcm.edu/genetics/index.cfm?pmid=21387. 2. The University of Chicago Genetic Services Laboratories https://dnatesting.uchicago.edu. 3. GeneDx http://www.genedx.com. 4. Transgenomic, 1-877-274-9432 http://www.transgenomic.com/labs/neurology.
- Labs: Serum vitamin E levels should be measured, and the vitamins administered to patients having deficient levels.
- Vitamin B1 and B12 levels in serum should be measured, and the vitamins administered to patients having deficient levels.
- Hypothyroidism is easily treated.
- The cerebrospinal fluid should be tested for a syphilitic infection in patients with progressive ataxia and other features of tabes dorsalis.
- Similarly, antibody titers for Lyme disease and Legionella should be measured and appropriate antibiotic therapy should be instituted in antibody-positive patients.
- Aminoacidopathies, leukodystrophies, urea-cycle abnormalities, and mitochondrial encephalomyopathies may produce ataxia, and some dietary or metabolic therapies are available for these disorders.
- The deleterious effects of phenytoin and alcohol on the cerebellum are well known, and these exposures should be avoided in patients with ataxia of any cause.
- Genetic testing: Genomic screening for CAG repeats in other families with autosomal dominant ataxia and vibratory and proprioceptive sensory loss have yielded another locus.
- Elevated serum alpha-fetoprotein and immunoglobulin deficiency are noted in AT.
- MRI findings: Cerebellar atrophy, spinal cord atrophy (Friedreich's), stable lesion on MRI/CT older than several months (vascular/demyelinating), malformation noted on MRI/CT (Chiari/Dandy-Walker), mass lesion on MRI/CT (abscess/tumor), positive for neoplasm on MRI/CT (glioma/metastatic tumor), positive HIV test and CD4+ cell count for AIDS (multifocal leukoencephalopathy).
- CSF: Supportive of acute viral infection, supportive of acute deficiency (alcoholic-nutritional), positive HIV test and CD4+ cell count for AIDS.
6.1 Diagnostic Algorithm¶
- Step 1: Assess onset (Acute, Subacute, Chronic) and symmetry (Symmetric vs Focal).
- Step 2: If Acute and Symmetric: Consider intoxication (alcohol, drugs, solvents), viral cerebellitis, postinfection syndrome.
- Step 3: If Subacute and Symmetric: Consider alcoholism + malnutrition (B1/B12 deficiency), hyponatremia, paraneoplastic syndromes, prion disorders.
- Step 4: If Chronic and Symmetric: Consider inherited ataxia, metabolic disorder, chronic infection (syphilis, Lyme).
- Step 5: If Focal and Ipsilateral: Consider vascular (infarction, hemorrhage), neoplastic (glioma, metastasis), infectious (abscess), demyelinating (MS), congenital (Chiari, Dandy-Walker).
- Step 6: Imaging: CT or MRI to reveal clinically significant processes. Look for herniation, mass lesions, stable lesions, malformations.
- Step 7: Labs: Vitamin E, B1, B12, Thyroid, Syphilis, Lyme, Legionella, HIV/CD4.
- Step 8: Genetic Testing: CAG repeats for SCAs, GAA repeats for Friedreich's, ATM gene for AT, mtDNA for mitochondrial ataxias.
- Step 9: Treatable Causes: Identify and treat hypothyroidism, vitamin deficiencies, infections, paraneoplastic syndromes, intoxication.
7. MANAGEMENT & TREATMENT¶
- The most important goal in management of patients with ataxia is to identify treatable disease entities.
- Mass lesions must be recognized promptly and treated appropriately.
- Autoimmune paraneoplastic disorders can often be identified by the clinical patterns of disease that they produce, measurement of specific autoantibodies, and uncovering the primary cancer; these disorders are often refractory to therapy, but some patients improve following removal of the tumor or immunotherapy.
- Ataxia with antigliadin antibodies and gluten-sensitive enteropathy may improve with a gluten-free diet.
- Malabsorption syndromes leading to vitamin E deficiency may lead to ataxia. The vitamin E deficiency form of Friedreich's ataxia must be considered, and serum vitamin E levels measured. Vitamin E therapy is indicated for these rare patients.
- Vitamin B1 and B12 levels in serum should be measured, and the vitamins administered to patients having deficient levels.
- Hypothyroidism is easily treated.
- The cerebrospinal fluid should be tested for a syphilitic infection in patients with progressive ataxia and other features of tabes dorsalis.
- Similarly, antibody titers for Lyme disease and Legionella should be measured and appropriate antibiotic therapy should be instituted in antibody-positive patients.
- Aminoacidopathies, leukodystrophies, urea-cycle abnormalities, and mitochondrial encephalomyopathies may produce ataxia, and some dietary or metabolic therapies are available for these disorders.
- The deleterious effects of phenytoin and alcohol on the cerebellum are well known, and these exposures should be avoided in patients with ataxia of any cause.
- There is no proven therapy for any of the autosomal dominant ataxias (SCA1 to SCA43).
- Omaveloxolone, a NRF2 agonist, is the only U.S. Food and Drug Administration–approved agent for Friedreich's ataxia.
- NRF2 is a transcription factor that regulates gene transcripts involved in mitochondrial energy production and addresses the root cause of mitochondrial dysfunction in Friedreich's ataxia.
- Iron chelators and antioxidant drugs are potentially harmful in Friedreich's patients because they may increase heart muscle injury.
- Acetazolamide can reduce the duration of symptoms of EA.
- At present, identification of an at-risk person's genotype, together with appropriate family and genetic counseling, can reduce the incidence of these cerebellar syndromes in future generations.
- Early detection of asymptomatic preclinical disease can reduce or eliminate the inherited form of ataxia in some families on a global, worldwide basis.
7.1 Pharmacologic Treatment¶
- Omaveloxolone: NRF2 agonist, FDA-approved for Friedreich's ataxia.
- Acetazolamide: Reduces duration of symptoms of Episodic Ataxia (EA).
- Vitamin E: Indicated for AVED and Abetalipoproteinemia.
- Vitamin B1 and B12: Administered to patients having deficient levels.
- Antibiotics: Appropriate antibiotic therapy for Lyme disease and Legionella in antibody-positive patients.
- Gluten-free diet: For ataxia with antigliadin antibodies and gluten-sensitive enteropathy.
- Thyroid Hormone: For hypothyroidism.
- Anticonvulsants: May be therapeutic for EA-1.
7.2 Genetic Counseling¶
- Identification of an at-risk person's genotype, together with appropriate family and genetic counseling, can reduce the incidence of these cerebellar syndromes in future generations.
- Genetic markers are now commercially available to precisely identify the genetic mutation for correct diagnosis and also for family planning.
- Early detection of asymptomatic preclinical disease can reduce or eliminate the inherited form of ataxia in some families on a global, worldwide basis.
8. PROGNOSIS & COMPLICATIONS¶
- Neurologic deficits invariably progress and lead to death from debilitation within 15 years of onset, especially in patients with types I and II disease (MJD).
- Usually, patients retain full intellectual function.
- The median age of death is 35 years (SCA7).
- Women have a significantly better prognosis than men (SCA7).
- Cardiac involvement occurs in 90% of patients (SCA7).
- Cardiomegaly, symmetric hypertrophy, murmurs, and conduction defects are reported (SCA7).
- Moderate intellectual disability or psychiatric syndromes are present in a small percentage of patients (SCA7).
- A high incidence (20%) of diabetes mellitus is found and is associated with insulin resistance and pancreatic β-cell dysfunction (SCA7).
- Musculoskeletal deformities are common and include pes cavus, pes equinovarus, and scoliosis (SCA7).
- MRI of the spinal cord shows atrophy (Friedreich's).
- The primary sites of pathology are the spinal cord, dorsal root ganglion cells, and the peripheral nerves (Friedreich's).
- Slight atrophy of the cerebellum and cerebral gyri may occur (Friedreich's).
- Sclerosis and degeneration occur predominantly in the spinocerebellar tracts, lateral corticospinal tracts, and posterior columns (Friedreich's).
- Degeneration of the glossopharyngeal, vagus, hypoglossal, and deep cerebellar nuclei is described (Friedreich's).
- The cerebral cortex is histologically normal except for loss of Betz cells in the precentral gyri (Friedreich's).
- The peripheral nerves are extensively involved, with a loss of large myelinated fibers (Friedreich's).
- Cardiac pathology consists of myocytic hypertrophy and fibrosis, focal vascular fibromuscular dysplasia with subintimal or medial deposition of periodic acid-Schiff (PAS)-positive material, and myocytopathy with unusual pleomorphic nuclei, and focal degeneration of nerves and cardiac ganglia (SCA7).
- There is an increased incidence of lymphomas, Hodgkin's disease, acute T-cell leukemias, and breast cancer (AT).
- Thymic hypoplasia with cellular and humoral (IgA and IgG2) immunodeficiencies, premature aging, and endocrine disorders such as type 1 diabetes mellitus are described (AT).
- There is a high incidence of recurrent pulmonary infections and neoplasms of the lymphatic and reticuloendothelial system in patients with AT.
8.1 Prognostic Factors¶
- MJD: Death from debilitation within 15 years of onset (Types I and II).
- SCA7: Median age of death is 35 years.
- SCA7: Women have a significantly better prognosis than men.
- SCA7: Cardiac involvement occurs in 90% of patients.
- AT: Increased incidence of lymphomas, Hodgkin's disease, acute T-cell leukemias, and breast cancer.
- Friedreich's: Progressive staggering gait, frequent falling, and titubation before 25 years of age.
9. SPECIAL CONSIDERATIONS¶
- Pregnancy: Not explicitly detailed in source text.
- Pediatrics: Juvenile patients have higher numbers of repeats (SCA1).
- Elderly: SCA27B occurs with a median age of onset of 60 years.
- Renal/Hepatic Impairment: Not explicitly detailed in source text.
- Immunocompromised: AIDS-related multifocal leukoencephalopathy (positive HIV test and CD4+ cell count for AIDS).
- Genetic Counseling: Identification of an at-risk person's genotype, together with appropriate family and genetic counseling, can reduce the incidence of these cerebellar syndromes in future generations.
10. KEY PEARLS & CLINICAL TRAPS¶
- Hypothyroidism must always be considered as a readily treatable and reversible form of gait ataxia.
- Prion disease treatment aims to reduce PrPC levels rather than directly eliminating PrPSc.
- Focal ataxia with headache, impaired consciousness, and ipsilateral cranial nerve palsies implies a space-occupying cerebellar lesion.
- Symmetric ataxia suggests genetic, metabolic, immune, or toxic etiology.
- Acute ataxia suggests intoxication or vascular disease.
- Chronic ataxia suggests inherited or metabolic disease.
- Vitamin E deficiency must be considered in Friedreich's ataxia.
- AT carries high risk for cancer (lymphomas, breast cancer).
- Omaveloxolone is the only FDA-approved agent for Friedreich's ataxia.
- Acetazolamide can reduce the duration of symptoms of EA.
WHAT TO LOOK FOR — DIAGNOSTIC CLUES¶
- Pathognomonic signs: Retinal pigmentary degeneration in SCA7 (blue-yellow color blindness, macular degeneration).
- Hallmark findings: Cerebellar atrophy on MRI (SCA1), Spinal cord atrophy (Friedreich's).
- Key features: Nystagmus, loss of fast saccadic eye movements, hypermetric and hypometric saccades, impairment of upward vertical gaze (SCA1, SCA3).
- Red flags: Sudden herniation (rostrally through the tentorium or caudal herniation of cerebellar tonsils through the foramen magnum) can occur and is usually devastating.
- Named eponymous signs: Romberg sign (removed in text context, but mentioned as a sign of ataxia).
- Lab signatures: Elevated serum alpha-fetoprotein and immunoglobulin deficiency in AT.
- Atypical presentations: Episodic ataxia (EA-1, EA-2) with brief episodes of ataxia induced by startle, sudden change in posture, or exercise.
- Clinical clues: Chronic unexplained cough is often associated with and may precede the onset of neurologic symptoms (CANVAS syndrome).
- Clinical pearls: Women have a significantly better prognosis than men (SCA7).
- Clinical pearls: Cardiac involvement occurs in 90% of patients (SCA7).
WHAT EXCLUDES THE DIAGNOSIS¶
- Not explicitly detailed in source text.
TABLES¶
- Table 450-1: Etiology of Cerebellar Ataxia.
Table 450-1: Etiology of Cerebellar Ataxia¶
- Symmetric and Progressive Signs: Acute (Hours to Days), Subacute (Days to Weeks), Chronic (Months to Years).
- Focal and Ipsilateral Cerebellar Signs: Acute (Hours to Days), Subacute (Days to Weeks), Chronic (Months to Years).
- Acute (Hours to Days) Symmetric: Intoxication (alcohol, lithium, phenytoin, barbiturates) (positive history and toxicology screen), Acute viral cerebellitis (CSF supportive of acute viral infection), Postinfection syndrome.
- Subacute (Days to Weeks) Symmetric: Intoxication (mercury, solvents, gasoline, glue), Cytotoxic chemotherapeutic drugs, Alcoholic-nutritional (vitamin B1 and B12 deficiency), Lyme disease, Phenytoin toxicity, Amiodarone.
- Chronic (Months to Years) Symmetric: Inherited diseases, Tabes dorsalis (tertiary syphilis), Hypothyroidism, AIDS-related multifocal leukoencephalopathy (positive HIV test and CD4+ cell count for AIDS), Stable gliosis secondary to vascular lesion or demyelinating plaque (stable lesion on MRI/CT older than several months), Congenital lesion (Chiari or Dandy-Walker malformations) (malformation noted on MRI/CT).
- Acute (Hours to Days) Focal: Vascular (cerebellar infarction, hemorrhage, or subdural hematoma), Infectious (cerebellar abscess) (mass lesion on MRI/CT, history in support of lesion).
- Subacute (Days to Weeks) Focal: Paraneoplastic syndrome, Antigliadin antibody syndrome.
- Chronic (Months to Years) Focal: Neoplastic (cerebellar glioma or metastatic tumor) (positive for neoplasm on MRI/CT), Demyelinating (multiple sclerosis) (history, CSF, and MRI are consistent), AIDS-related multifocal leukoencephalopathy (positive HIV test and CD4+ cell count for AIDS), Stable gliosis secondary to vascular lesion or demyelinating plaque (stable lesion on MRI/CT older than several months), Congenital lesion (Chiari or Dandy-Walker malformations) (malformation noted on MRI/CT).
- Abbreviations: CSF, cerebrospinal fluid; CT, computed tomography; MRI, magnetic resonance imaging.
Table 1 — Table 450-1 Etiology of Cerebellar Ataxia¶
| Symmetric and Progressive Signs | Focal and Ipsilateral Cerebellar Signs |
|---|---|
| Acute (Hours to Days) | Acute (Hours to Days) |
| Intoxication: alcohol, lithium, phenytoin, barbiturates (positive history and toxicology screen) | Vascular: cerebellar infarction, hemorrhage, or subdural hematoma |
| Acute viral cerebellitis (CSF supportive of acute viral infection) | Infectious: cerebellar abscess (mass lesion on MRI/CT, history in support of lesion) |
| Postinfection syndrome | Neoplastic: cerebellar glioma or metastatic tumor (positive for neoplasm on MRI/CT) |
| Subacute (Days to Weeks) | Subacute (Days to Weeks) |
| Intoxication: mercury, solvents, gasoline, glue | Paraneoplastic syndrome |
| Cytotoxic chemotherapeutic drugs | Antigliadin antibody syndrome |
| Alcoholic-nutritional (vitamin B1 and B12 deficiency) | Demyelinating: multiple sclerosis (history, CSF, and MRI are consistent) |
| Lyme disease | AIDS-related multifocal leukoencephalopathy (positive HIV test and CD4+ cell count for AIDS) |
| Phenytoin toxicity | Stable gliosis secondary to vascular lesion or demyelinating plaque (stable lesion on MRI/CT older than several months) |
| Amiodarone | Congenital lesion: Chiari or Dandy-Walker malformations (malformation noted on MRI/CT) |
| Chronic (Months to Years) | Chronic (Months to Years) |
| Inherited diseases | Stable gliosis secondary to vascular lesion or demyelinating plaque (stable lesion on MRI/CT older than several months) |
| Tabes dorsalis (tertiary syphilis) | Congenital lesion: Chiari or Dandy-Walker malformations (malformation noted on MRI/CT) |
| Hypothyroidism | |
| AIDS-related multifocal leukoencephalopathy (positive HIV test and CD4+ cell count for AIDS) |
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 450-1 Sagittal magnetic resonance imaging (MRI) of the brain of a 60-year- old man with gait ataxia and dysarthria due to spinocerebellar ataxia type 1 (SCA1), illustrating cerebellar atrophy (arrows). (Reproduced with permission from RN Rosenberg, P Khemani, in RN Rosenberg, JM Pascual [eds]: Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed. London, Elsevier, 2015.) — Sagittal magnetic resonance imaging (MRI) of the brain of a 60-year-old man with gait ataxia and dysarthria due to spinocerebellar ataxia type 1 (SCA1), illustrating cerebellar atrophy (arrows).
Figure 2¶
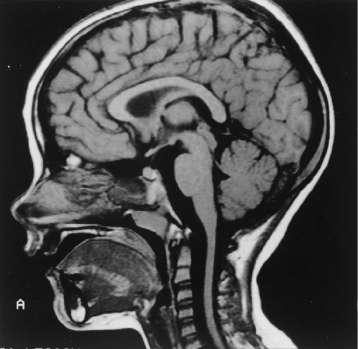
Caption: FIGURE 450-2 Sagittal magnetic resonance imaging (MRI) of the brain and spinal cord of a patient with Friedreich’s ataxia, demonstrating spinal cord atrophy. of (Reproduced with permission from RN Rosenberg, P Khemani, in RN Rosenberg, JM Pascual [eds]: Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed. London, Elsevier, 2015.) — Sagittal magnetic resonance imaging (MRI) of the brain and spinal cord of a patient with Friedreich's ataxia, demonstrating spinal cord atrophy.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.