Cystic Fibrosis¶
Chapter 302 | Part 7: Disorders of the Respiratory System · Part 7 – Respiratory Disorders
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- CF is an autosomal recessive exocrinopathy caused by mutations in the CFTR gene.
- Diagnosis requires two disease-causing CFTR variants and/or sweat chloride ≥60 mEq/L.
- F508del is the most common mutation (~85% of defective alleles in the US).
- CFTR modulators (e.g., ivacaftor, TCT) are highly effective for specific genotypes (HEMT).
- Pulmonary exacerbations require aggressive inpatient antibiotic therapy (aminoglycoside + beta-lactam).
- Lung transplantation is indicated for FEV1 <30% predicted or severe decline.
- CF carrier status predisposes to non-CF bronchiectasis and chronic rhinosinusitis.
- Sweat chloride ≥60 mEq/L is the cardinal diagnostic test; hyperviscosity of sweat is not a clinical feature.
- Survival has improved significantly with modulators, with median survival >60 years in the US.
- CFTR modulators are FDA approved for specific genotypes; access is restricted by cost and approval status.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 Respiratory Manifestations
- 1.2 Pancreatic Findings
- 1.3 Additional Organ System Damage
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 CFTR Mutation Classes
- 4. CLINICAL FEATURES
- 4.1 Respiratory Manifestations
- 4.2 Pancreatic Findings
- 4.3 Additional Organ System Damage
- 5. DIFFERENTIAL DIAGNOSIS
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Diagnostic Criteria
- 6.2 Diagnostic Algorithm
- 7. MANAGEMENT & TREATMENT
- 7.1 CFTR Modulators
- 7.2 Chronic Outpatient Management
- 7.3 Pulmonary Exacerbation
- 7.4 Lung Transplantation
- 8. PROGNOSIS & COMPLICATIONS
- 8.1 Survival & Longevity
- 8.2 Complications
- 9. SPECIAL CONSIDERATIONS
- 9.1 Aging & Comorbidities
- 9.2 Reproductive Health
- 10. KEY PEARLS & CLINICAL TRAPS
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🖼 Figure | Extrusion of mucus secretion onto the epithelial surface of reticulum airways in... |
| 2 | 🖼 Figure | Extrusion of mucus secretion onto the epithelial surface of reticulum airways in... |
1. DEFINITION & OVERVIEW¶
- Cystic fibrosis (CF) is an autosomal recessive exocrinopathy affecting multiple epithelial tissues.
- The gene product responsible for CF is the cystic fibrosis transmembrane conductance regulator (CFTR).
- CFTR is an integral membrane protein that functions as an epithelial anion channel.
- CFTR serves as an anion channel in the apical (luminal) plasma membranes of epithelial cells and regulates volume and composition of exocrine secretion.
- Harrison's defines this as: 'Cystic fibrosis (CF) is an autosomal recessive exocrinopathy affecting multiple epithelial tissues.'
- The ~1480-amino-acid molecule encodes a passive conduit for chloride and bicarbonate transport across plasma membranes of epithelial tissues.
- Direction of ion flow is dependent on the electrochemical driving force.
- Gating of CFTR involves conformational cycling between an open and closed configuration and is augmented by hydrolysis of adenosine triphosphate (ATP).
- Anion flux mediated by CFTR does not involve active transport against a concentration gradient but utilizes the energy provided from ATP hydrolysis as a central feature of ion channel mechanochemistry and gating.
- CFTR is situated in the apical plasma membranes of acinar and other epithelial cells where it regulates the amount and composition of secretion by exocrine glands.
- In numerous epithelia, chloride and bicarbonate release via CFTR is followed passively by flow of water through other pathways, aiding mobilization and clearance of exocrine products.
- Along respiratory mucosa, CFTR is necessary to provide sufficient depth of the periciliary fluid layer (PCL), allowing normal ciliary extension and mucociliary transport.
- CFTR-deficient airway cells exhibit depleted PCL, causing ciliary collapse and failure to clear overlying mucus.
- In airway submucosal glands, CFTR is expressed in acini and may participate both in the formation of mucus and extrusion of glandular secretion onto the airway surface.
- In other exocrine glands characterized by abrogated mucus transport (e.g., pancreatic acini and ducts, as well as bile canaliculi, and intestinal tissues), similar pathogenic mechanisms have been implicated.
- Failure of this mechanism disrupts normal hydration and transport of glandular secretion—and is viewed as a proximate cause of obstruction, with concomitant tissue injury.
1.1 Respiratory Manifestations¶
- The major morbidity and mortality associated with CF is attributable to pulmonary compromise.
- CF respiratory secretions are characterized by copious hyperviscous and adherent secretions that obstruct small and medium-sized airways.
- CF respiratory secretions are exceedingly difficult to clear.
- A complex bacterial flora that includes Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa (among other pathogens) is routinely cultured from CF sputum.
- Robust pulmonary inflammation in the setting of inspissated mucus and chronic infection leads to collateral tissue injury and further aggravates respiratory decline.
- Organisms such as P. aeruginosa exhibit a stereotypic mode of pathogenesis; a sentinel early colonization event often engenders lifelong pulmonary infection by the same genetic strain.
- Over a period of many years, P. aeruginosa evolves in CF lungs to adopt a mucoid phenotype (attributable to release of alginate exoproduct) that confers selective advantage for the pathogen and poor prognosis for the host.
- Radiographic evidence of sinusitis occurs in most patients with CF and can be associated with microorganisms similar to those recovered from lower airways, suggesting that CF sinus involvement may serve as a reservoir for bacterial seeding of the lung.
- The CF airway is characterized by an aggressive, unrelenting, neutrophilic inflammatory response with release of proteases and oxidants leading to airway remodeling and bronchiectasis.
- Intense pulmonary inflammation is largely driven by chronic respiratory pathogens, although some measure of inflammatory responsiveness may occur very early in the disease and prior to bacterial infection.
- Macrophages and other cells resident in CF lungs augment elaboration of proinflammatory cytokines, which contribute to innate and adaptive immune reactivity.
- CFTR-dependent abnormalities of airway surface fluid composition (e.g., low pH) have been reported as contributors to impaired bacterial killing in CF lungs.
- The role of CFTR activity as a direct mediator of cellular immune hyperresponsiveness and/or pulmonary remodeling represent important areas of investigation.
1.2 Pancreatic Findings¶
- The complete name of the disease, cystic fibrosis of the pancreas, refers to profound tissue destruction of the exocrine pancreas.
- Pathology includes fibrotic scarring and/or fatty replacement, cyst formation, loss of acinar tissue, and ablation of normal pancreatic architecture.
- As in the lung, tenacious exocrine secretions (sometimes termed concretions) obstruct pancreatic ducts and impair production and flow of digestive enzymes to the duodenum.
- The sequelae of exocrine pancreatic insufficiency include chronic malabsorption, poor growth, fat-soluble vitamin deficiency, high levels of blood immunoreactive trypsinogen (a test used in newborn screening), low levels of fecal elastase-1, and loss of pancreatic islet cell mass.
- CF-related diabetes mellitus is a manifestation in >30% of adults with the disease (and likely multifactorial in nature).
- CF-related diabetes is attributable to progressive destruction/dysfunction of the endocrine pancreas and, in some cases, insulin resistance or other features.
1.3 Additional Organ System Damage¶
- As in CF lung and pancreas, thick and inspissated secretions compromise numerous exocrine tissues.
- Obstruction of intrahepatic bile ducts and parenchymal fibrosis are observed, with multilobular cirrhosis in 4–15% of patients with CF and significant hepatic insufficiency as a resulting manifestation among many adults.
- Hepatic steatosis, focal biliary fibrosis, and portal hypertension are other well-described findings of CF liver disease.
- Contents of the intestinal lumen are hyperviscous and often difficult to excrete, leading to meconium ileus (a clinical presentation in many newborns with CF) or distal intestinal obstructive syndrome in older individuals.
- Men typically exhibit complete involution of the vas deferens (despite functioning spermatogenesis), and ~99% of males with CF are infertile (i.e., unable to conceive children without fertilization).
- Abnormalities of female reproductive tract secretions are likely contributors to a higher incidence of infertility among women with the disease.
2. EPIDEMIOLOGY¶
- Although the disease has traditionally been viewed as most common among whites (~1 in 3300 live U.S. births), there is growing concern that CF has been underdiagnosed in certain parts of the world.
- CF in regions such as Eastern Europe, Latin America, Asia, and India—as well as among minority populations in the United States—is sometimes associated with rare variants that are less well defined and/or poorly characterized.
- In many clinical settings, therefore, enhanced attention to diagnosis is needed, particularly when specialized DNA testing and sweat chloride measurements are unavailable and patients with CF may be missed.
- Failure to identify CF properly in early childhood has important implications, since nutritional and other therapies at a young age are believed to promote quality of life and increase longevity.
- As one example, median survival among individuals with CF is 60 years in the United States).
- The less favorable prognosis is attributable in part to lack of widespread diagnostic capabilities (i.e., newborn screening, sweat testing, and genetic analysis tailored to ethnic background), together with insufficient access to leading-edge, interdisciplinary treatment.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- DNA sequencing from CF patients (and others) worldwide has revealed >2000 allelic mutations in CFTR; several hundred of these are well characterized as disease-causing variants.
- Distinguishing the single nucleotide transversions or other polymorphisms with causal relevance can sometimes present a significant challenge.
- The CFTR2 resource (www.cftr2.org/) helps delineate gene variants with a clear etiologic role.
- CFTR defects known to elicit disease have been categorized based on molecular mechanism.
- For example, the common F508del mutation (nomenclature denotes omission of a single phenylalanine residue [F] at CFTR position 508) leads to a protein folding abnormality recognized by cellular quality control pathways.
- CFTR encoding F508del retains partial ion channel function, but protein maturation is arrested in vitro in the endoplasmic reticulum, and CFTR fails to arrive at the plasma membrane.
- Instead, F508del CFTR is misrouted and undergoes endoplasmic reticulum–associated degradation via the proteasome.
- CFTR mutations that disrupt protein maturation in this manner are termed class II defects and are by far the most common genetic abnormalities.
- F508del alone accounts for ~85% of defective CFTR alleles in the United States.
- Other gene defects include CFTR ion channels properly trafficked to the apical cell surface but unable to open and/or gate.
- Such channel proteins include G551D (a glycine to aspartic acid replacement at CFTR position 551), which abrogates transport of Cl– or HCO– (a class III abnormality).
- Individuals with at least one G551D allele represent ~4% of patients with CF.
- CFTR nonsense mutations such as G542X, R553X, or W1282X (premature termination codon replaces glycine, arginine, or tryptophan at positions 542, 553, or 1282, respectively) are among the common class I defects, in addition to large deletions or other major disruptions of the gene.
- The W1282X mutation, for example, is prevalent among individuals of Ashkenazi descent and a predominant CF genotype in Israel.
- Additional categories of CFTR mutation include impairment of the ion channel pore (class IV), RNA splicing (class V), and increased plasma membrane turnover (class VI).
- The magnitude of transepithelial sodium reabsorption in CF airways, which helps control periciliary fluid depth and composition, is strongly influenced by CFTR and has represented a molecular target for experimental intervention in the past.
3.1 CFTR Mutation Classes¶
- Class I: Absence of synthesis.
- Class II: Defective protein maturation and premature degradation.
- Class III: Disordered gating/regulation, such as diminished adenosine triphosphate (ATP) binding and hydrolysis.
- Class IV: Defective conductance through the ion channel pore.
- Class V: A reduced number of CFTR transcripts due to a promoter or splicing abnormality.
- Class VI: Accelerated turnover from the cell surface.
Table 1 — Table 302-1. Categories of CFTR Mutations¶
| Class | Defect Description | Example Mutations | Frequency/Notes |
|---|---|---|---|
| Class I | Absence of synthesis | Nonsense mutations (e.g., G542X, R553X, W1282X) | Common class I defects; W1282X prevalent in Ashkenazi/Israel |
| Class II | Defective protein maturation and premature degradation | F508del (omission of phenylalanine at position 508) | Most common genetic abnormalities; accounts for ~85% of defective alleles in US |
| Class III | Disordered gating/regulation | G551D (glycine to aspartic acid at position 551) | Abrogates transport of Cl– or HCO–; ~4% of patients |
| Class IV | Defective conductance through the ion channel pore | Various pore mutations | Impairment of ion channel pore |
| Class V | Reduced number of CFTR transcripts | Promoter or splicing abnormality | Reduced number of CFTR transcripts |
| Class VI | Accelerated turnover from the cell surface | Various turnover mutations | Increased plasma membrane turnover |
4. CLINICAL FEATURES¶
- CF classically presented in childhood with chronic productive cough, malabsorption including steatorrhea, and failure to thrive.
- During the past decade, newborn screening has led to most CF diagnoses, with confirmation through CFTR mutation analysis and/or sweat electrolyte measurements as cardinal tests.
- The findings of disease-causing CFTR variants on both CFTR alleles and/or sweat chloride ≥60 mEq/L, together with characteristic respiratory or other exocrine manifestations, are sufficient for confirming a diagnosis of CF.
- DNA-based evaluation typically surveys numerous disease-associated mutations; panels that identify several hundred CFTR variants are available through a variety of public health laboratories or commercial sources.
- Alternatively, complete CFTR DNA testing, or exonic sequencing together with analysis of splice junctions and key regulatory elements, can be obtained.
- Sweat electrolytes following pilocarpine iontophoresis comprise an essential diagnostic element, with levels of chloride markedly elevated in CF compared to non-CF individuals.
- The sweat test result is highly specific and served as a mainstay of diagnosis for decades prior to availability of CFTR genotyping.
- Notably, hyperviscosity of eccrine sweat is not a clinical feature of the disease.
- Sweat ducts function to reabsorb chloride from a primary sweat secretion produced by the glandular coil.
- Malfunction of CFTR leads to diminished chloride uptake from the ductular lumen, and sweat emerges on the skin with elevated levels of chloride.
- Several 'severe' defects that impair CFTR activity (including F508del, G551D, and nonsense alleles) are predictive of pancreatic insufficiency, which is evident in ~80% of those with the disease.
- In general, and with regard to projecting respiratory outcome for an individual patient, genotype has been of limited value for predicting the rate of clinical decline, respiratory prognosis, or longevity.
- A spectrum of CFTR-related conditions with features resembling classic CF has been well described.
- In addition to multiorgan involvement, forme frustes, such as isolated congenital bilateral absence of the vas deferens or pancreatitis (without other organ system findings), are strongly associated with CFTR mutations in at least one allele.
- CF carrier status also predisposes to both non-CF bronchiectasis and chronic rhinosinusitis, indicating a contribution of relative CFTR deficiency in these prevalent illnesses.
- Although CF is a classic monogenic disease, the importance of non-CFTR gene modifiers and proteins that regulate ion flux, inflammatory pathways, and airway remodeling has been appreciated as influencing clinical course.
4.1 Respiratory Manifestations¶
- The major morbidity and mortality associated with CF is attributable to pulmonary compromise.
- CF respiratory secretions are characterized by copious hyperviscous and adherent secretions that obstruct small and medium-sized airways.
- CF respiratory secretions are exceedingly difficult to clear.
- A complex bacterial flora that includes Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa (among other pathogens) is routinely cultured from CF sputum.
- Robust pulmonary inflammation in the setting of inspissated mucus and chronic infection leads to collateral tissue injury and further aggravates respiratory decline.
- Organisms such as P. aeruginosa exhibit a stereotypic mode of pathogenesis; a sentinel early colonization event often engenders lifelong pulmonary infection by the same genetic strain.
- Over a period of many years, P. aeruginosa evolves in CF lungs to adopt a mucoid phenotype (attributable to release of alginate exoproduct) that confers selective advantage for the pathogen and poor prognosis for the host.
- Radiographic evidence of sinusitis occurs in most patients with CF and can be associated with microorganisms similar to those recovered from lower airways, suggesting that CF sinus involvement may serve as a reservoir for bacterial seeding of the lung.
- The CF airway is characterized by an aggressive, unrelenting, neutrophilic inflammatory response with release of proteases and oxidants leading to airway remodeling and bronchiectasis.
- Intense pulmonary inflammation is largely driven by chronic respiratory pathogens, although some measure of inflammatory responsiveness may occur very early in the disease and prior to bacterial infection.
- Macrophages and other cells resident in CF lungs augment elaboration of proinflammatory cytokines, which contribute to innate and adaptive immune reactivity.
- CFTR-dependent abnormalities of airway surface fluid composition (e.g., low pH) have been reported as contributors to impaired bacterial killing in CF lungs.
- The role of CFTR activity as a direct mediator of cellular immune hyperresponsiveness and/or pulmonary remodeling represent important areas of investigation.
4.2 Pancreatic Findings¶
- The complete name of the disease, cystic fibrosis of the pancreas, refers to profound tissue destruction of the exocrine pancreas.
- Pathology includes fibrotic scarring and/or fatty replacement, cyst formation, loss of acinar tissue, and ablation of normal pancreatic architecture.
- As in the lung, tenacious exocrine secretions (sometimes termed concretions) obstruct pancreatic ducts and impair production and flow of digestive enzymes to the duodenum.
- The sequelae of exocrine pancreatic insufficiency include chronic malabsorption, poor growth, fat-soluble vitamin deficiency, high levels of blood immunoreactive trypsinogen (a test used in newborn screening), low levels of fecal elastase-1, and loss of pancreatic islet cell mass.
- CF-related diabetes mellitus is a manifestation in >30% of adults with the disease (and likely multifactorial in nature).
- CF-related diabetes is attributable to progressive destruction/dysfunction of the endocrine pancreas and, in some cases, insulin resistance or other features.
4.3 Additional Organ System Damage¶
- As in CF lung and pancreas, thick and inspissated secretions compromise numerous exocrine tissues.
- Obstruction of intrahepatic bile ducts and parenchymal fibrosis are observed, with multilobular cirrhosis in 4–15% of patients with CF and significant hepatic insufficiency as a resulting manifestation among many adults.
- Hepatic steatosis, focal biliary fibrosis, and portal hypertension are other well-described findings of CF liver disease.
- Contents of the intestinal lumen are hyperviscous and often difficult to excrete, leading to meconium ileus (a clinical presentation in many newborns with CF) or distal intestinal obstructive syndrome in older individuals.
- Men typically exhibit complete involution of the vas deferens (despite functioning spermatogenesis), and ~99% of males with CF are infertile (i.e., unable to conceive children without fertilization).
- Abnormalities of female reproductive tract secretions are likely contributors to a higher incidence of infertility among women with the disease.
5. DIFFERENTIAL DIAGNOSIS¶
- CF carrier status also predisposes to both non-CF bronchiectasis and chronic rhinosinusitis, indicating a contribution of relative CFTR deficiency in these prevalent illnesses.
- Although CF is a classic monogenic disease, the importance of non-CFTR gene modifiers and proteins that regulate ion flux, inflammatory pathways, and airway remodeling has been appreciated as influencing clinical course.
- A spectrum of CFTR-related conditions with features resembling classic CF has been well described.
- In addition to multiorgan involvement, forme frustes, such as isolated congenital bilateral absence of the vas deferens or pancreatitis (without other organ system findings), are strongly associated with CFTR mutations in at least one allele.
- CF carrier status also predisposes to both non-CF bronchiectasis and chronic rhinosinusitis, indicating a contribution of relative CFTR deficiency in these prevalent illnesses.
6. INVESTIGATIONS & DIAGNOSIS¶
- CF classically presented in childhood with chronic productive cough, malabsorption including steatorrhea, and failure to thrive.
- During the past decade, newborn screening has led to most CF diagnoses, with confirmation through CFTR mutation analysis and/or sweat electrolyte measurements as cardinal tests.
- The findings of disease-causing CFTR variants on both CFTR alleles and/or sweat chloride ≥60 mEq/L, together with characteristic respiratory or other exocrine manifestations, are sufficient for confirming a diagnosis of CF.
- DNA-based evaluation typically surveys numerous disease-associated mutations; panels that identify several hundred CFTR variants are available through a variety of public health laboratories or commercial sources.
- Alternatively, complete CFTR DNA testing, or exonic sequencing together with analysis of splice junctions and key regulatory elements, can be obtained.
- Sweat electrolytes following pilocarpine iontophoresis comprise an essential diagnostic element, with levels of chloride markedly elevated in CF compared to non-CF individuals.
- The sweat test result is highly specific and served as a mainstay of diagnosis for decades prior to availability of CFTR genotyping.
- Notably, hyperviscosity of eccrine sweat is not a clinical feature of the disease.
- Sweat ducts function to reabsorb chloride from a primary sweat secretion produced by the glandular coil.
- Malfunction of CFTR leads to diminished chloride uptake from the ductular lumen, and sweat emerges on the skin with elevated levels of chloride.
6.1 Diagnostic Criteria¶
- Confirmation requires disease-causing CFTR variants on both CFTR alleles and/or sweat chloride ≥60 mEq/L.
- Characteristic respiratory or other exocrine manifestations are also sufficient for confirming a diagnosis of CF.
- DNA-based evaluation typically surveys numerous disease-associated mutations.
- Panels that identify several hundred CFTR variants are available through a variety of public health laboratories or commercial sources.
- Alternatively, complete CFTR DNA testing, or exonic sequencing together with analysis of splice junctions and key regulatory elements, can be obtained.
Table 2 — Table 6.1. Diagnostic Criteria for Cystic Fibrosis¶
| Diagnostic Test | Threshold/Result | Significance |
|---|---|---|
| Sweat Chloride (Pilocarpine Iontophoresis) | ≥60 mEq/L | Highly specific; mainstay of diagnosis for decades |
| CFTR Genotyping | Disease-causing variants on both alleles | Sufficient for confirming diagnosis |
| Clinical Features | Characteristic respiratory or other exocrine manifestations | Sufficient for confirming diagnosis (with genetic/sweat findings) |
6.2 Diagnostic Algorithm¶
- Step 1: Clinical suspicion based on chronic productive cough, malabsorption including steatorrhea, and failure to thrive.
- Step 2: Newborn screening has led to most CF diagnoses.
- Step 3: Confirmation through CFTR mutation analysis and/or sweat electrolyte measurements as cardinal tests.
- Step 4: DNA-based evaluation typically surveys numerous disease-associated mutations.
- Step 5: Alternatively, complete CFTR DNA testing, or exonic sequencing together with analysis of splice junctions and key regulatory elements, can be obtained.
- Step 6: The findings of disease-causing CFTR variants on both CFTR alleles and/or sweat chloride ≥60 mEq/L, together with characteristic respiratory or other exocrine manifestations, are sufficient for confirming a diagnosis of CF.
7. MANAGEMENT & TREATMENT¶
- Standard care for patients with CF is intensive, with outpatient regimens that include exogenous pancreatic enzymes taken with meals, nutritional supplementation, anti-inflammatory medication, bronchodilators, and chronic or periodic dosing of oral or aerosolized antibiotics (e.g., as maintenance therapy for patients with P. aeruginosa).
- Recombinant DNAse aerosols (degrade DNA strands that contribute to mucus viscosity) and nebulized hypertonic saline or mannitol (augment PCL depth, activate mucociliary clearance, and mobilize inspissated airway secretions) have traditionally been administered.
- Chest physiotherapy several times each day is routinely used as a means to promote clearance of airway mucus, although rigorous evidence for clinical benefit is limited.
- Among adults with CF, intestinal malabsorption, chronic inflammation, and endocrine abnormalities can lead to poor bone mineralization requiring treatment with vitamin D, calcium, and other measures.
- The time, complexity, and expense of home care are considerable and take a significant toll on patients and their families.
- Important CF longitudinal trials are reevaluating the need for interventions, such as recombinant DNAse, and examining optimal guidelines for frequency of outpatient clinic visits, home monitoring, sputum culture surveillance, and other aspects of clinical care in the era of highly effective modulators.
- Chronic sequelae of CF have received continued attention following the advent of HEMT, particularly since patients with established lung disease given TCT or other formulations continue to exhibit respiratory infection and inflammation despite substantial clinical improvement.
- Certain disease hallmarks, such as CF sinus involvement, are palliated by HEMT, and both exocrine pancreatic function and glucose tolerance have been shown to benefit from modulators in specific clinical settings.
- Annual testing for diabetes mellitus continues to be recommended for adults with CF.
- The impact of CFTR modulation has not been fully characterized for other extrapulmonary manifestations of the disease.
- Improved treatments that address ongoing nutritional deficits, hepatic and endocrine abnormalities, mucostasis, or additional features that persist despite modulators remain a priority.
- Sexual and reproductive health have become areas of considerable interest, since pregnancies are markedly increased among women on HEMT.
- Understanding potential relationship(s) between clinical depression, overall mental health, and chronic modulator therapy remains an important future and emerging topic.
- Pulmonary Exacerbation: Severe CF respiratory exacerbation is commonly managed by hospital admission for parenteral antibiotics and frequent chest physiotherapy directed against (often multidrug-resistant) bacterial pathogens.
- Aggressive intervention in this setting can restore a large component of lung function, but ongoing and cumulative loss of pulmonary reserve has traditionally reflected the natural history of the disease.
- Poor prognostic indicators such as sputum culture containing Burkholderia cepacia complex, Stenotrophomonas maltophilia, Achromobacter, mucoid P. aeruginosa, or atypical mycobacteria are rigorously monitored in the CF patient population.
- Methicillin-resistant S. aureus in CF lungs may also be associated with less favorable outcomes.
- As noted above, while HEMT may diminish bacterial density in sputum samples, infection commonly persists despite modulator treatment.
- Typical inpatient antibiotic coverage includes combination drug therapy with an aminoglycoside and β-lactam (if Pseudomonas is present) for ~14 days.
- Maximal improvement in lung function is often achieved by 8–10 days, although optimal duration of therapy is a subject of continuing investigation.
- Many families elect parenteral antibiotic treatment at home, but additional studies are needed to evaluate specific drug combinations, duration of therapy, and home versus inpatient management.
- Other CF respiratory sequelae that may require hospitalization include hemoptysis and pneumothorax.
- Hypersensitivity to Aspergillus (allergic bronchopulmonary aspergillosis) occurs in ~5% of individuals with CF and should be suspected in the absence of a beneficial response to aggressive inpatient antibiotics.
- Lung Transplantation: For end-stage CF pulmonary failure, transplantation is a viable therapeutic option with median survival >9 years among adults with the disease.
- Determining optimal timing for surgery presents a substantial challenge in patients with severe respiratory compromise, particularly since the rate of continued functional decline, as well as individualized mortality risk from transplantation, can be difficult to predict.
- FEV measurements 60 years in the United States).
- The less favorable prognosis is attributable in part to lack of widespread diagnostic capabilities (i.e., newborn screening, sweat testing, and genetic analysis tailored to ethnic background), together with insufficient access to leading-edge, interdisciplinary treatment.
- Efforts to apply state-of-the-art management to underdiagnosed and underserved CF patient populations will help improve outcomes and mitigate CF health disparities in the future.
7.1 CFTR Modulators¶
- Potentiation of Mutant CFTR Gating: A major effort directed toward high-throughput analysis of large compound libraries resulted in identification of effective new CFTR 'modulator' therapies for CF.
- The first approved compound in this class, ivacaftor, robustly potentiates CFTR channel opening and stimulates ion transport.
- Ivacaftor overcomes the G551D CFTR gating defect, and individuals carrying this mutation exhibit pronounced improvement in lung function, respiratory outcomes (fewer hospital admissions for pulmonary exacerbation), weight gain, and other clinical benefits.
- Ivacaftor has been deemed 'highly effective modulator therapy' (HEMT) for G551D-related CF and leads to substantial reduction of sweat chloride.
- Prior to ivacaftor, no clinical intervention of any sort had been shown to normalize the CF sweat phenotype.
- In addition to G551D, ivacaftor is approved in the United States for 96 other CFTR variants.
- Multiyear treatment analysis indicates durable respiratory palliation.
- The drug has been viewed as a harbinger of a new era for CF therapeutics directed toward addressing fundamental causes of the disease.
- Correction of the F508del Processing Abnormality: Lumacaftor and tezacaftor, two U.S. Food and Drug Administration (FDA)-approved 'corrector' molecules that repair CFTR misfolding (as distinct from CFTR gating 'potentiators' such as ivacaftor), partially overcome the F508del biogenesis defect.
- The drugs also promote cell surface localization of many other class II CFTR mutations.
- A different corrector molecule, elexacaftor, operates by a distinct mechanism of action and is FDA approved in combination with tezacaftor and ivacaftor for patients with CF encoding at least one F508del variant (irrespective of the second CFTR allele), as well as numerous less common CF mutations.
- This triple combination therapy (TCT) has been projected to benefit >90% of individuals with the disease.
- Marked enhancement of forced expiratory volume in 1 s (FEV), fewer respiratory exacerbations, improved quality of life, and diminished sweat chloride have all been demonstrated in patients following TCT, leading to designation as HEMT.
- For example, among individuals carrying one F508del together with a CFTR minimal function variant, TCT led to improved FEV (% predicted) by ~14% over a 4- to 24-week treatment period.
- Monitoring liver function of patients on TCT and attention to pharmacologic interactions, including effects mediated by CYP3A, are required.
- Personalized Molecular Therapies: Based on the large number of disease-causing CFTR mutations, together with the ability to group these into molecular categories (Fig. 302-2), CF has been deemed a condition ideally suited for personalized (i.e., mechanistically tailored) drug treatment.
- That being said, many CFTR variants clearly exhibit multiple molecular abnormalities (across more than one mechanistic subclass), and modulator compounds can therefore provide benefit across numerous disease subcategories.
- CFTR drug discovery—while highly successful—might, therefore, be viewed as less 'personalized' or 'precise' than originally envisioned.
- Moreover, clinical data indicate that a subset of individuals with F508del respond poorly to TCTs.
- Other Challenges Involving CFTR Modulators and Progress: The high cost of modulator compounds has often restricted third-party reimbursement to include only the specific genotypes for which FDA or other regulatory approval has been obtained.
- As a consequence, access to potentially efficacious modulators among patients with very rare CFTR defects and off-label prescribing are largely precluded.
- Moreover, clinical trials intended to expand the drug label can be difficult to arrange based on small numbers of patients carrying a particular ultra-rare variant.
- In vitro models rigorously shown to predict clinical modulator benefit have proven useful in this setting (e.g., studies of primary airway and other well-validated epithelial monolayers, or organoid-type cultures) and represent a potential means to gain FDA approval or insurance reimbursement for those with uncommon CFTR abnormalities.
- Toward Nucleotide-Based Therapeutics: The high cost of modulator compounds has often restricted third-party reimbursement to include only the specific genotypes for which FDA or other regulatory approval has been obtained.
- As a consequence, access to potentially efficacious modulators among patients with very rare CFTR defects and off-label prescribing are largely precluded.
- Moreover, clinical trials intended to expand the drug label can be difficult to arrange based on small numbers of patients carrying a particular ultra-rare variant.
- In vitro models rigorously shown to predict clinical modulator benefit have proven useful in this setting (e.g., studies of primary airway and other well-validated epithelial monolayers, or organoid-type cultures) and represent a potential means to gain FDA approval or insurance reimbursement for those with uncommon CFTR abnormalities.
- CF drug discovery is emblematic of what might be accomplished in other refractory inherited conditions using an approach grounded in molecular etiology and unbiased compound library screening.
- Beyond CFTR modulators, genetic manipulation (e.g., CFTR gene transfer, DNA editing) and airway progenitor cell engraftment comprise experimental approaches that may be less dependent on a specific (i.e., personalized) pathogenic mechanism.
- For example, efficient, safe, and durable delivery of wild-type CFTR using viral (e.g., adeno-associated), lipid nanoparticle, or other vehicles represents a potential means to address diverse molecular abnormalities, independent of the responsible CFTR mutation(s).
- In that context, rescue of secretory epithelial progenitor cells has been emphasized, with newer technologies such as single-cell RNA-seq or spatial transcriptomics available to track successful nucleotide-based transduction.
- Approaches to genetic correction are of particular urgency for patients with forms of CF unresponsive to CFTR modulation, such as disease caused by premature truncation codons or disruptive splice site abnormalities.
Table 3 — Table 7.1. CFTR Modulator Therapies¶
| Drug | Class | Mechanism | Indication | Monitoring/Notes |
|---|---|---|---|---|
| Ivacaftor | Potentiator | Potentiates CFTR channel opening and stimulates ion transport | G551D-related CF and 96 other CFTR variants | HEMT for G551D; reduces sweat chloride |
| Lumacaftor | Corrector | Repairs CFTR misfolding; promotes cell surface localization | F508del biogenesis defect | Distinct from gating potentiators |
| Tezacaftor | Corrector | Repairs CFTR misfolding; promotes cell surface localization | F508del biogenesis defect | Distinct from gating potentiators |
| Elexacaftor | Corrector | Distinct mechanism of action | In combination with tezacaftor and ivacaftor | FDA approved for patients with CF encoding at least one F508del variant |
| Triple Combination Therapy (TCT) | Corrector + Potentiator | Combination of elexacaftor, tezacaftor, and ivacaftor | Patients with CF encoding at least one F508del variant | Benefit >90% of individuals; improved FEV, fewer exacerbations |
7.2 Chronic Outpatient Management¶
- Standard care for patients with CF is intensive, with outpatient regimens that include exogenous pancreatic enzymes taken with meals, nutritional supplementation, anti-inflammatory medication, bronchodilators, and chronic or periodic dosing of oral or aerosolized antibiotics (e.g., as maintenance therapy for patients with P. aeruginosa).
- Recombinant DNAse aerosols (degrade DNA strands that contribute to mucus viscosity) and nebulized hypertonic saline or mannitol (augment PCL depth, activate mucociliary clearance, and mobilize inspissated airway secretions) have traditionally been administered.
- Chest physiotherapy several times each day is routinely used as a means to promote clearance of airway mucus, although rigorous evidence for clinical benefit is limited.
- Among adults with CF, intestinal malabsorption, chronic inflammation, and endocrine abnormalities can lead to poor bone mineralization requiring treatment with vitamin D, calcium, and other measures.
- The time, complexity, and expense of home care are considerable and take a significant toll on patients and their families.
- Important CF longitudinal trials are reevaluating the need for interventions, such as recombinant DNAse, and examining optimal guidelines for frequency of outpatient clinic visits, home monitoring, sputum culture surveillance, and other aspects of clinical care in the era of highly effective modulators.
- Chronic sequelae of CF have received continued attention following the advent of HEMT, particularly since patients with established lung disease given TCT or other formulations continue to exhibit respiratory infection and inflammation despite substantial clinical improvement.
- Certain disease hallmarks, such as CF sinus involvement, are palliated by HEMT, and both exocrine pancreatic function and glucose tolerance have been shown to benefit from modulators in specific clinical settings.
- Annual testing for diabetes mellitus continues to be recommended for adults with CF.
- The impact of CFTR modulation has not been fully characterized for other extrapulmonary manifestations of the disease.
- Improved treatments that address ongoing nutritional deficits, hepatic and endocrine abnormalities, mucostasis, or additional features that persist despite modulators remain a priority.
- Sexual and reproductive health have become areas of considerable interest, since pregnancies are markedly increased among women on HEMT.
- Understanding potential relationship(s) between clinical depression, overall mental health, and chronic modulator therapy remains an important future and emerging topic.
7.3 Pulmonary Exacerbation¶
- Severe CF respiratory exacerbation is commonly managed by hospital admission for parenteral antibiotics and frequent chest physiotherapy directed against (often multidrug-resistant) bacterial pathogens.
- Aggressive intervention in this setting can restore a large component of lung function, but ongoing and cumulative loss of pulmonary reserve has traditionally reflected the natural history of the disease.
- Poor prognostic indicators such as sputum culture containing Burkholderia cepacia complex, Stenotrophomonas maltophilia, Achromobacter, mucoid P. aeruginosa, or atypical mycobacteria are rigorously monitored in the CF patient population.
- Methicillin-resistant S. aureus in CF lungs may also be associated with less favorable outcomes.
- As noted above, while HEMT may diminish bacterial density in sputum samples, infection commonly persists despite modulator treatment.
- Typical inpatient antibiotic coverage includes combination drug therapy with an aminoglycoside and β-lactam (if Pseudomonas is present) for ~14 days.
- Maximal improvement in lung function is often achieved by 8–10 days, although optimal duration of therapy is a subject of continuing investigation.
- Many families elect parenteral antibiotic treatment at home, but additional studies are needed to evaluate specific drug combinations, duration of therapy, and home versus inpatient management.
- Other CF respiratory sequelae that may require hospitalization include hemoptysis and pneumothorax.
- Hypersensitivity to Aspergillus (allergic bronchopulmonary aspergillosis) occurs in ~5% of individuals with CF and should be suspected in the absence of a beneficial response to aggressive inpatient antibiotics.
7.4 Lung Transplantation¶
- For end-stage CF pulmonary failure, transplantation is a viable therapeutic option with median survival >9 years among adults with the disease.
- Determining optimal timing for surgery presents a substantial challenge in patients with severe respiratory compromise, particularly since the rate of continued functional decline, as well as individualized mortality risk from transplantation, can be difficult to predict.
- FEV measurements <30% predicted, together with an assortment of other clinical parameters (e.g., hospitalization frequency, need for supplemental oxygen, modulator treatment), are employed as thresholds for transplant referral, although patients with conditions such as significant pulmonary hypertension may merit consultation at higher FEV.
- In general, evaluation for pulmonary transplant has often been underutilized among patients with CF.
- The decision is best approached based on early input from providers specializing in both CF clinical management and transplant medicine.
8. PROGNOSIS & COMPLICATIONS¶
- Although the disease has traditionally been viewed as most common among whites (~1 in 3300 live U.S. births), there is growing concern that CF has been underdiagnosed in certain parts of the world, including regions such as Eastern Europe, Latin America, Asia, and India.
- CF in these areas—as well as among minority populations in the United States—is sometimes associated with rare variants that are less well defined and/or poorly characterized.
- In many clinical settings, therefore, enhanced attention to diagnosis is needed, particularly when specialized DNA testing and sweat chloride measurements are unavailable and patients with CF may be missed.
- Failure to identify CF properly in early childhood has important implications, since nutritional and other therapies at a young age are believed to promote quality of life and increase longevity.
- As one example, median survival among individuals with CF is 60 years in the United States).
- The less favorable prognosis is attributable in part to lack of widespread diagnostic capabilities (i.e., newborn screening, sweat testing, and genetic analysis tailored to ethnic background), together with insufficient access to leading-edge, interdisciplinary treatment.
- Efforts to apply state-of-the-art management to underdiagnosed and underserved CF patient populations will help improve outcomes and mitigate CF health disparities in the future.
- As a direct result of advances in basic research, modulator and other therapies have transformed CF from a disease that historically led to death in early childhood to a condition with frequent survival well into the fifth decade of life and beyond.
- Initiating modulator treatment in young children with CF is expected to further promote longevity by forestalling pulmonary damage, although this assertion will require formal testing in patients on long-term HEMT.
- Care of aging individuals with CF (in which older patients with the disease experience obesity, cancer, vascular, and other age-related comorbidities commonly observed in the general population) has become an emerging priority.
- As modulatory therapies advance, standardized approaches to treatment will be essential.
- Well-defined protocols for general CF management are already widely established, including guidance regarding hospital admission, antibiotic regimens, nutritional intervention, periodicity of diagnostic tests, and other clinical parameters.
- These recommendations are implemented by specialized CF care centers and similarly accredited programs.
- Such measures have led to markedly improved pulmonary function, weight gain, body mass index, and other clinical endpoints among patients with the disease.
- The same approach is being applied to help optimize care of individuals given new CFTR modulators, as well as older patients with CF.
- Standardized protocols for CF therapy can be accessed at https://www.cff.org/managing-cf/new-era-cf-care-possible-changes or through a number of excellent reviews.
8.1 Survival & Longevity¶
- Median survival among individuals with CF is 60 years in the United States).
- The less favorable prognosis is attributable in part to lack of widespread diagnostic capabilities (i.e., newborn screening, sweat testing, and genetic analysis tailored to ethnic background), together with insufficient access to leading-edge, interdisciplinary treatment.
- As a direct result of advances in basic research, modulator and other therapies have transformed CF from a disease that historically led to death in early childhood to a condition with frequent survival well into the fifth decade of life and beyond.
- Initiating modulator treatment in young children with CF is expected to further promote longevity by forestalling pulmonary damage, although this assertion will require formal testing in patients on long-term HEMT.
8.2 Complications¶
- Chronic sequelae of CF have received continued attention following the advent of HEMT, particularly since patients with established lung disease given TCT or other formulations continue to exhibit respiratory infection and inflammation despite substantial clinical improvement.
- Certain disease hallmarks, such as CF sinus involvement, are palliated by HEMT, and both exocrine pancreatic function and glucose tolerance have been shown to benefit from modulators in specific clinical settings.
- Annual testing for diabetes mellitus continues to be recommended for adults with CF.
- The impact of CFTR modulation has not been fully characterized for other extrapulmonary manifestations of the disease.
- Improved treatments that address ongoing nutritional deficits, hepatic and endocrine abnormalities, mucostasis, or additional features that persist despite modulators remain a priority.
- Sexual and reproductive health have become areas of considerable interest, since pregnancies are markedly increased among women on HEMT.
- Understanding potential relationship(s) between clinical depression, overall mental health, and chronic modulator therapy remains an important future and emerging topic.
- Other CF respiratory sequelae that may require hospitalization include hemoptysis and pneumothorax.
- Hypersensitivity to Aspergillus (allergic bronchopulmonary aspergillosis) occurs in ~5% of individuals with CF and should be suspected in the absence of a beneficial response to aggressive inpatient antibiotics.
- Poor prognostic indicators such as sputum culture containing Burkholderia cepacia complex, Stenotrophomonas maltophilia, Achromobacter, mucoid P. aeruginosa, or atypical mycobacteria are rigorously monitored in the CF patient population.
- Methicillin-resistant S. aureus in CF lungs may also be associated with less favorable outcomes.
- Among adults with CF, intestinal malabsorption, chronic inflammation, and endocrine abnormalities can lead to poor bone mineralization requiring treatment with vitamin D, calcium, and other measures.
9. SPECIAL CONSIDERATIONS¶
- Care of aging individuals with CF (in which older patients with the disease experience obesity, cancer, vascular, and other age-related comorbidities commonly observed in the general population) has become an emerging priority.
- As modulatory therapies advance, standardized approaches to treatment will be essential.
- Well-defined protocols for general CF management are already widely established, including guidance regarding hospital admission, antibiotic regimens, nutritional intervention, periodicity of diagnostic tests, and other clinical parameters.
- These recommendations are implemented by specialized CF care centers and similarly accredited programs.
- Such measures have led to markedly improved pulmonary function, weight gain, body mass index, and other clinical endpoints among patients with the disease.
- The same approach is being applied to help optimize care of individuals given new CFTR modulators, as well as older patients with CF.
- Standardized protocols for CF therapy can be accessed at https://www.cff.org/managing-cf/new-era-cf-care-possible-changes or through a number of excellent reviews.
- Sexual and reproductive health have become areas of considerable interest, since pregnancies are markedly increased among women on HEMT.
- Understanding potential relationship(s) between clinical depression, overall mental health, and chronic modulator therapy remains an important future and emerging topic.
- Improved treatments that address ongoing nutritional deficits, hepatic and endocrine abnormalities, mucostasis, or additional features that persist despite modulators remain a priority.
9.1 Aging & Comorbidities¶
- Care of aging individuals with CF (in which older patients with the disease experience obesity, cancer, vascular, and other age-related comorbidities commonly observed in the general population) has become an emerging priority.
- As modulatory therapies advance, standardized approaches to treatment will be essential.
- Well-defined protocols for general CF management are already widely established, including guidance regarding hospital admission, antibiotic regimens, nutritional intervention, periodicity of diagnostic tests, and other clinical parameters.
- These recommendations are implemented by specialized CF care centers and similarly accredited programs.
- Such measures have led to markedly improved pulmonary function, weight gain, body mass index, and other clinical endpoints among patients with the disease.
- The same approach is being applied to help optimize care of individuals given new CFTR modulators, as well as older patients with CF.
- Standardized protocols for CF therapy can be accessed at https://www.cff.org/managing-cf/new-era-cf-care-possible-changes or through a number of excellent reviews.
9.2 Reproductive Health¶
- Sexual and reproductive health have become areas of considerable interest, since pregnancies are markedly increased among women on HEMT.
- Men typically exhibit complete involution of the vas deferens (despite functioning spermatogenesis), and ~99% of males with CF are infertile (i.e., unable to conceive children without fertilization).
- Abnormalities of female reproductive tract secretions are likely contributors to a higher incidence of infertility among women with the disease.
10. KEY PEARLS & CLINICAL TRAPS¶
- CF classically presented in childhood with chronic productive cough, malabsorption including steatorrhea, and failure to thrive.
- During the past decade, newborn screening has led to most CF diagnoses, with confirmation through CFTR mutation analysis and/or sweat electrolyte measurements as cardinal tests.
- The findings of disease-causing CFTR variants on both CFTR alleles and/or sweat chloride ≥60 mEq/L, together with characteristic respiratory or other exocrine manifestations, are sufficient for confirming a diagnosis of CF.
- DNA-based evaluation typically surveys numerous disease-associated mutations; panels that identify several hundred CFTR variants are available through a variety of public health laboratories or commercial sources.
- Alternatively, complete CFTR DNA testing, or exonic sequencing together with analysis of splice junctions and key regulatory elements, can be obtained.
- Sweat electrolytes following pilocarpine iontophoresis comprise an essential diagnostic element, with levels of chloride markedly elevated in CF compared to non-CF individuals.
- The sweat test result is highly specific and served as a mainstay of diagnosis for decades prior to availability of CFTR genotyping.
- Notably, hyperviscosity of eccrine sweat is not a clinical feature of the disease.
- Sweat ducts function to reabsorb chloride from a primary sweat secretion produced by the glandular coil.
- Malfunction of CFTR leads to diminished chloride uptake from the ductular lumen, and sweat emerges on the skin with elevated levels of chloride.
- Several 'severe' defects that impair CFTR activity (including F508del, G551D, and nonsense alleles) are predictive of pancreatic insufficiency, which is evident in ~80% of those with the disease.
- In general, and with regard to projecting respiratory outcome for an individual patient, genotype has been of limited value for predicting the rate of clinical decline, respiratory prognosis, or longevity.
- A spectrum of CFTR-related conditions with features resembling classic CF has been well described.
- In addition to multiorgan involvement, forme frustes, such as isolated congenital bilateral absence of the vas deferens or pancreatitis (without other organ system findings), are strongly associated with CFTR mutations in at least one allele.
- CF carrier status also predisposes to both non-CF bronchiectasis and chronic rhinosinusitis, indicating a contribution of relative CFTR deficiency in these prevalent illnesses.
- Although CF is a classic monogenic disease, the importance of non-CFTR gene modifiers and proteins that regulate ion flux, inflammatory pathways, and airway remodeling has been appreciated as influencing clinical course.
- The magnitude of transepithelial sodium reabsorption in CF airways, which helps control periciliary fluid depth and composition, is strongly influenced by CFTR and has represented a molecular target for experimental intervention in the past.
- CF drug discovery is emblematic of what might be accomplished in other refractory inherited conditions using an approach grounded in molecular etiology and unbiased compound library screening.
- Beyond CFTR modulators, genetic manipulation (e.g., CFTR gene transfer, DNA editing) and airway progenitor cell engraftment comprise experimental approaches that may be less dependent on a specific (i.e., personalized) pathogenic mechanism.
- For example, efficient, safe, and durable delivery of wild-type CFTR using viral (e.g., adeno-associated), lipid nanoparticle, or other vehicles represents a potential means to address diverse molecular abnormalities, independent of the responsible CFTR mutation(s).
- In that context, rescue of secretory epithelial progenitor cells has been emphasized, with newer technologies such as single-cell RNA-seq or spatial transcriptomics available to track successful nucleotide-based transduction.
- Approaches to genetic correction are of particular urgency for patients with forms of CF unresponsive to CFTR modulation, such as disease caused by premature truncation codons or disruptive splice site abnormalities.
- The high cost of modulator compounds has often restricted third-party reimbursement to include only the specific genotypes for which FDA or other regulatory approval has been obtained.
- As a consequence, access to potentially efficacious modulators among patients with very rare CFTR defects and off-label prescribing are largely precluded.
- Moreover, clinical trials intended to expand the drug label can be difficult to arrange based on small numbers of patients carrying a particular ultra-rare variant.
- In vitro models rigorously shown to predict clinical modulator benefit have proven useful in this setting (e.g., studies of primary airway and other well-validated epithelial monolayers, or organoid-type cultures) and represent a potential means to gain FDA approval or insurance reimbursement for those with uncommon CFTR abnormalities.
- Newborn screening for CF is universal throughout the United States and Canadian provinces, Australia, New Zealand, and most of Europe, and facilitates early intervention.
- Although the disease has traditionally been viewed as most common among whites (~1 in 3300 live U.S. births), there is growing concern that CF has been underdiagnosed in certain parts of the world, including regions such as Eastern Europe, Latin America, Asia, and India.
- CF in these areas—as well as among minority populations in the United States—is sometimes associated with rare variants that are less well defined and/or poorly characterized.
- In many clinical settings, therefore, enhanced attention to diagnosis is needed, particularly when specialized DNA testing and sweat chloride measurements are unavailable and patients with CF may be missed.
- Failure to identify CF properly in early childhood has important implications, since nutritional and other therapies at a young age are believed to promote quality of life and increase longevity.
- As one example, median survival among individuals with CF is 60 years in the United States).
- The less favorable prognosis is attributable in part to lack of widespread diagnostic capabilities (i.e., newborn screening, sweat testing, and genetic analysis tailored to ethnic background), together with insufficient access to leading-edge, interdisciplinary treatment.
- Efforts to apply state-of-the-art management to underdiagnosed and underserved CF patient populations will help improve outcomes and mitigate CF health disparities in the future.
- Pulmonary Exacerbation: Severe CF respiratory exacerbation is commonly managed by hospital admission for parenteral antibiotics and frequent chest physiotherapy directed against (often multidrug-resistant) bacterial pathogens.
- Aggressive intervention in this setting can restore a large component of lung function, but ongoing and cumulative loss of pulmonary reserve has traditionally reflected the natural history of the disease.
- Poor prognostic indicators such as sputum culture containing Burkholderia cepacia complex, Stenotrophomonas maltophilia, Achromobacter, mucoid P. aeruginosa, or atypical mycobacteria are rigorously monitored in the CF patient population.
- Methicillin-resistant S. aureus in CF lungs may also be associated with less favorable outcomes.
- As noted above, while HEMT may diminish bacterial density in sputum samples, infection commonly persists despite modulator treatment.
- Typical inpatient antibiotic coverage includes combination drug therapy with an aminoglycoside and β-lactam (if Pseudomonas is present) for ~14 days.
- Maximal improvement in lung function is often achieved by 8–10 days, although optimal duration of therapy is a subject of continuing investigation.
- Many families elect parenteral antibiotic treatment at home, but additional studies are needed to evaluate specific drug combinations, duration of therapy, and home versus inpatient management.
- Other CF respiratory sequelae that may require hospitalization include hemoptysis and pneumothorax.
- Hypersensitivity to Aspergillus (allergic bronchopulmonary aspergillosis) occurs in ~5% of individuals with CF and should be suspected in the absence of a beneficial response to aggressive inpatient antibiotics.
- Lung Transplantation: For end-stage CF pulmonary failure, transplantation is a viable therapeutic option with median survival >9 years among adults with the disease.
- Determining optimal timing for surgery presents a substantial challenge in patients with severe respiratory compromise, particularly since the rate of continued functional decline, as well as individualized mortality risk from transplantation, can be difficult to predict.
- FEV measurements 60 years in the United States).
- The less favorable prognosis is attributable in part to lack of widespread diagnostic capabilities (i.e., newborn screening, sweat testing, and genetic analysis tailored to ethnic background), together with insufficient access to leading-edge, interdisciplinary treatment.
- Efforts to apply state-of-the-art management to underdiagnosed and underserved CF patient populations will help improve outcomes and mitigate CF health disparities in the future.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶
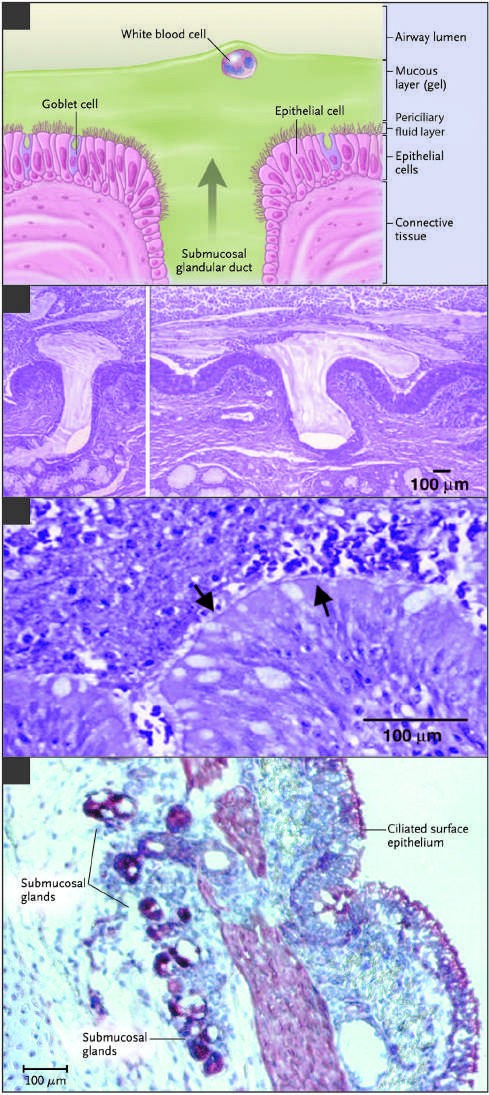
Caption: FIGURE 302-1 Extrusion of mucus secretion onto the epithelial surface of reticulum airways in cystic fibrosis (CF). A. Schematic of the surface epithelium and supporting glandular structure of the human airway. B. The submucosal glands of a patient with CF are filled with mucus, and mucopurulent debris overlies the airway surfaces, essentially burying the epithelium. C. A higher magnification view of a mucus plug tightly adhering to the airway surface, with arrows indicating Nucleus Class I Class V the interface between infected and inflamed secretions and the underlying epithelium to which the secretions adhere. (Both B and C were stained with hematoxylin and eosin, with the colors modified to highlight structures.) Infected secretions obstruct airways and, over time, dramatically disrupt the normal architecture of the lung. D. CFTR is expressed in surface epithelium and serous — Figure 302-1: Extrusion of mucus secretion onto the epithelial surface of airways in cystic fibrosis (CF). A. Schematic of the surface epithelium and supporting glandular structure of the human airway. B. The submucosal glands of a patient with CF are filled with mucus, and mucopurulent debris overlies the airway surfaces, essentially burying the epithelium. C. A higher magnification view of a mucus plug tightly adhering to the airway surface, with arrows indicating the interface between infected and inflamed secretions and the underlying epithelium. D. CFTR expression in surface epithelium and serous cells at the base of submucosal glands in a porcine lung sample.
Figure 2¶

Caption: FIGURE 302-1 Extrusion of mucus secretion onto the epithelial surface of reticulum airways in cystic fibrosis (CF). A. Schematic of the surface epithelium and supporting glandular structure of the human airway. B. The submucosal glands of a patient with CF are filled with mucus, and mucopurulent debris overlies the airway surfaces, essentially burying the epithelium. C. A higher magnification view of a mucus plug tightly adhering to the airway surface, with arrows indicating Nucleus Class I Class V the interface between infected and inflamed secretions and the underlying epithelium to which the secretions adhere. (Both B and C were stained with hematoxylin and eosin, with the colors modified to highlight structures.) Infected secretions obstruct airways and, over time, dramatically disrupt the normal architecture of the lung. D. CFTR is expressed in surface epithelium and serous — Figure 302-2: Categories of CFTR mutations. Classes of defects in the CFTR gene include the absence of synthesis (class I); defective protein maturation and premature degradation (class II); disordered gating/regulation (class III); defective conductance through the ion channel pore (class IV); a reduced number of CFTR transcripts due to a promoter or splicing abnormality (class V); and accelerated turnover from the cell surface (class VI).
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.