Chapter 402: Sex Development¶
Part 12: Endocrinology and Metabolism · Part 12 – Endocrinology & Metabolism
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- In the newborn period, approximately 1 in 5000 babies undergo investigation due to atypical or ambiguous genitalia.
- Sex development is divided into three components: chromosomal sex, gonadal sex, and phenotypic sex.
- Klinefelter syndrome (47,XXY) incidence is at least 1 in 1000 men, but ~75% of cases are not diagnosed.
- Turner syndrome affects approximately 1 in 2500 women; ~1 in 2500 women have a 45,X karyotype.
- The existence of Y chromosome material in individuals with Turner syndrome increases the risk for germ cell tumors.
- Complete gonadal dysgenesis (Swyer's syndrome) is associated with streak gonads and a 46,XY karyotype.
- Androgen insensitivity syndrome (AIS) is X-linked; only 46,XY offspring are affected.
- Salt-losing crisis is a life-threatening presentation associated with congenital adrenal hyperplasia (CAH).
- Gonadectomy is traditionally advised for Turner syndrome if Y chromosome material exists due to tumor risk.
- Recombinant growth hormone is used to increase adult height in Turner syndrome.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 Components of Sex Development
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Genetic Regulation of Gonadal Development
- 3.2 Androgen Synthesis Pathways
- 4. CLINICAL FEATURES
- 4.1 Presentation by Life Stage
- 4.2 Associated Clinical Features by Disorder
- 5. DIFFERENTIAL DIAGNOSIS
- 5.1 Genetic Causes of 46,XY DSDs
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Diagnostic Algorithm
- 7. MANAGEMENT & TREATMENT
- 7.1 Hormone Replacement Therapy
- 7.2 Surgical and Procedural Management
- 8. PROGNOSIS & COMPLICATIONS
- 8.1 Long-term Monitoring
- 9. SPECIAL CONSIDERATIONS
- 10. KEY PEARLS & CLINICAL TRAPS
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🖼 Figure | Sex development can be divided into three major components: chromosomal sex, gonadal... |
| 2 | 🖼 Figure | Simplified overview of glucocorticoid and androgen synthesis pathways |
| 3 | 🖼 Figure | The genetic regulation of gonadal development |
| 4 | 🖼 Figure | The genetic regulation of gonadal development |
| 5 | 🖼 Figure | The genetic regulation of gonadal development |
1. DEFINITION & OVERVIEW¶
- Sex development begins in utero but continues into young adulthood with the achievement of sexual maturity and reproductive capability.
- The major determinants of sex development can be divided into three components: chromosomal sex, gonadal sex (sex determination), and phenotypic sex (sex differentiation).
- Variations at each of these stages can result in differences (or disorders) of sex development (DSDs).
- Previous terms such as intersex and hermaphrodite were changed by the 2006 Consensus Statement to disorder of sex development and ovotesticular DSD.
- The term “disorder” is often considered negative and stigmatizing and thus has shifted toward difference of sex development, but no term is universally accepted.
- Care of individuals with DSDs should be holistic, often involving medical, psychosocial, and urogynecologic expertise.
- An experienced multidisciplinary team is important for counseling, planning appropriate investigations, discussing long-term well-being, supporting parents, and providing clear communication about the diagnosis and management options.
1.1 Components of Sex Development¶
- Chromosomal sex: Defined by a karyotype, describes the X and/or Y chromosome complement (46,XY; 46,XX) established at the time of fertilization.
- Gonadal sex: Refers to the histologic and functional characteristics of gonadal tissue as testis or ovary.
- Phenotypic sex: Refers to the structures of the external and internal genitalia and secondary sex characteristics.
2. EPIDEMIOLOGY¶
- In the newborn period, ~1 in 5000 babies undergo investigation because of atypical or ambiguous genitalia.
- Klinefelter syndrome (KS) has an incidence of at least 1 in 1000 men, but ~75% of cases are not diagnosed.
- Turner syndrome affects approximately 1 in 2500 women.
- Approximately one-half of women with TS have a 45,X karyotype, ~20% have 45,X/46,XX mosaicism, and the remainder have structural abnormalities of the X chromosome, or Y chromosome material.
- Isolated hypospadias occurs in ~1 in 250 males.
- Unilateral undescended testes (cryptorchidism) affect >3% of boys at birth.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- Testis development is initiated by expression of the gene SRY (sex-determining region on the Y chromosome) (from ~42 days after conception).
- Disruption of SRY prevents testis development in 46,XY individuals.
- Translocation of SRY in 46,XX individuals induces testis development and a male phenotype.
- SRY regulates SOX9 (SRY-related HMG-box gene 9), leading to expression of a cascade of genes involved in testis development.
- Ovarian development is not a “passive” process. Many specific genes are expressed during early ovary development, some of which may repress testis development (e.g., WNT4, R-spondin-1).
- Germ cells also develop in a sex dimorphic manner. In the developing ovary, primordial germ cells (PGCs) show marked proliferation and enter meiosis, whereas they undergo mitotic arrest in the developing testis.
- Approximately 7 million germ cells are present in the fetal ovary toward the end of the second trimester, and 1 million remain at birth.
- Only 400 are ovulated during a woman’s reproductive life span.
- Defects in enzymes involved in androgen synthesis result in underandrogenization of the 46,XY fetus.
- Defects in the pathway that regulates androgen synthesis cause underandrogenization of the 46,XY fetus.
- Pathogenic variants in the androgen receptor cause resistance to androgen (testosterone, DHT) action or the androgen insensitivity syndrome (AIS).
3.1 Genetic Regulation of Gonadal Development¶
- SRY: Sex-determining region on the Y chromosome.
- SOX9: SRY-related HMG-box gene 9.
- WNT4: Wingless-type MMTV integration site 4.
- RSPO1: R-spondin 1.
- FOXL2: Forkhead transcription factor L2.
- WT1: Wilms’ tumor–related gene 1.
- SF1: Steroidogenic factor 1 (also known as NR5A1).
- DAX1: Dosage sensitive sex-reversal, adrenal hypoplasia congenita on the X chromosome, gene 1.
3.2 Androgen Synthesis Pathways¶
- ACTH stimulates adrenal steroidogenesis.
- LH stimulates testicular steroidogenesis.
- StAR (Steroidogenic acute regulatory protein) facilitates cholesterol transport into mitochondria.
- CYP11A1 (Cholesterol side-chain cleavage enzyme) converts cholesterol to pregnenolone.
- CYP17A1 (17α-hydroxylase/17,20-lyase) converts pregnenolone to 17-hydroxypregnenolone.
- CYP21A2 (21-hydroxylase) converts 17-hydroxypregnenolone to 11-deoxycortisol.
- CYP11B1 (11-hydroxylase) converts 11-deoxycortisol to cortisol.
- CYP19A1 (Aromatase) converts testosterone to estradiol.
- SRD5A2 (5α-reductase) converts testosterone to dihydrotestosterone (DHT).
4. CLINICAL FEATURES¶
- KS is most commonly characterized by small testes, infertility, gynecomastia, tall stature/increased leg length, and hypogonadism in phenotypic males.
- At birth, most infants with KS do not have clinical features, although there are higher rates of cryptorchidism and hypospadias.
- Most patients present in puberty with arrested pubertal development caused by testicular insufficiency.
- TS is characterized by female-typical external genitalia, short stature, hypergonadotropic hypogonadism, infertility, ovarian insufficiency and other phenotypic features.
- Infants may present with lymphedema, nuchal folds, low hairline, or left-sided cardiac defects.
- Children may present with unexplained growth failure or delayed puberty.
- Ovotesticular DSD usually presents with atypical genitalia at birth and sometimes breast development, cyclical hematuria, and/or phallic development at puberty.
- 45,X/46,XY mosaicism can vary considerably; some have a predominantly female phenotype, most have a male phenotype.
- Persistent müllerian duct syndrome is the presence of a uterus in an otherwise phenotypic male.
4.1 Presentation by Life Stage¶
- Prenatal: Karyotype-phenotype discordance, atypical genitalia.
- Neonatal: Salt-losing crisis, hernia, androgenization.
- Childhood: Poor growth, associated features.
- Puberty: Androgenization, estrogenization, absent puberty.
- Post-puberty: Amenorrhea.
- Adult: Infertility.
Table 1 — Table 402-2 Presentation of Differences of Sex Development (DSD) at Different Stages of Life¶
| PRESENTATION | FEATURES | PROFESSIONAL | EXAMPLES |
|---|---|---|---|
| Prenatal | Karyotype-phenotype discordance | Obstetrician; fetal medicine | Many |
| Neonatal | Atypical genitalia | Obstetrician; neonatal medicine | Many |
| Neonatal | Salt-losing crisis | Pediatrician | CAH (CYP21A2) |
| Neonatal | Hernia | Surgeon | CAH (CYP21A2, CYP11B1) |
| Neonatal | Androgenization | Endocrinologist | CAH (CYP21A2, CYP11B1) |
| Childhood | Poor growth | Pediatrician | Turner, 45,X/46,XY |
| Childhood | Associated features | Oncologist/nephrologist | Wilms’ tumor |
| Puberty | Androgenization | Endocrinologist | 17β-HSD, 5α-reductase, SF1 |
| Puberty | Estrogenization | Endocrinologist | Ovotestis |
| Puberty | Absent puberty | Endocrinologist | Gonadal dysgenesis, CAH (CYP17A1), Turner |
| Post-puberty | Amenorrhea | Gynecologist | CAIS |
| Adult | Infertility | Andrologist | Klinefelter, 45,X/46,XY, SF1 |
4.2 Associated Clinical Features by Disorder¶
- Klinefelter syndrome: Small testes, azoospermia, decreased facial and axillary hair, decreased libido, tall stature and increased leg length, decreased penile length, increased risk of breast tumors, thromboembolic disease, learning difficulties, anxiety, speech delay and decreased verbal IQ, obesity, diabetes mellitus, metabolic syndrome, varicose veins, hypothyroidism, systemic lupus erythematosus, epilepsy.
- Turner syndrome: Infancy (lymphedema, web neck, shield chest, low-set hairline, cardiac defects and coarctation of the aorta, urinary tract malformations, and horseshoe kidney), Childhood (short stature, cubitus valgus, short neck, short fourth metacarpals, hypoplastic nails, micrognathia, scoliosis, otitis media and sensorineural hearing loss, ptosis and amblyopia, multiple nevi and keloid formation, autoimmune thyroid disease, visuospatial learning difficulties), Adulthood (absent puberty and primary amenorrhea, hypertension, obesity, dyslipidemia, impaired glucose tolerance and insulin resistance, autoimmune thyroid disease, cardiovascular disease, aortic root dilation, osteoporosis, inflammatory bowel disease, chronic hepatic dysfunction, increased risk of colon cancer, hearing loss).
- 45,X/46,XY mosaicism: Short stature, increased risk of gonadal tumors, some Turner syndrome features.
- Ovotesticular DSD: Possible increased risk of gonadal tumors.
Table 2 — Table 402-3 Possible Associated Clinical Features of Sex Chromosome Variations¶
| DISORDER | COMMON CHROMOSOMAL COMPLEMENT | GONAD | EXTERNAL GENITALIA | INTERNAL GENITALIA | BREAST DEVELOPMENT | CLINICAL FEATURES |
|---|---|---|---|---|---|---|
| Klinefelter syndrome | 47,XXY or 46,XY/47,XXY | Hyalinized testes | Male | Male | Increased incidence of gynecomastia | Small testes, azoospermia, decreased facial and axillary hair, decreased libido, tall stature and increased leg length, decreased penile length, increased risk of breast tumors, thromboembolic disease, learning difficulties, anxiety, speech delay and decreased verbal IQ, obesity, diabetes mellitus, metabolic syndrome, varicose veins, hypothyroidism, systemic lupus erythematosus, epilepsy |
| Turner syndrome | 45,X or 45,X/46,XX | Streak gonad or immature ovary | Female | Hypoplastic female | Immature female | Infancy: lymphedema, web neck, shield chest, low-set hairline, cardiac defects and coarctation of the aorta, urinary tract malformations, and horseshoe kidney; Childhood: short stature, cubitus valgus, short neck, short fourth metacarpals, hypoplastic nails, micrognathia, scoliosis, otitis media and sensorineural hearing loss, ptosis and amblyopia, multiple nevi and keloid formation, autoimmune thyroid disease, visuospatial learning difficulties; Adulthood: absent puberty and primary amenorrhea, hypertension, obesity, dyslipidemia, impaired glucose tolerance and insulin resistance, autoimmune thyroid disease, cardiovascular disease, aortic root dilation, osteoporosis, inflammatory bowel disease, chronic hepatic dysfunction, increased risk of colon cancer, hearing loss |
| 45,X/46,XY mosaicism | 45,X/46,XY | Testis or streak gonad | Variable | Variable | Usually male | Short stature, increased risk of gonadal tumors, some Turner syndrome features |
| Ovotesticular DSD | 46,XX/46,XY | Testis and ovary or ovotestis | Variable | Variable | Gynecomastia | Possible increased risk of gonadal tumors |
5. DIFFERENTIAL DIAGNOSIS¶
- DSDs can present at other ages and to a range of health professionals, including with absent puberty, primary amenorrhea, or androgenization in the teen years.
- Other forms of gonadal dysfunction (e.g., Klinefelter syndrome [KS], Turner syndrome [TS]) often are diagnosed later in life by internists.
- Karyotype is a useful starting investigation for diagnosis with basic biochemical profiling.
- High-throughput genetic testing can help in reaching a definitive diagnosis.
- Differential diagnosis includes disruption of testis development, disruption of androgen synthesis, disruption of androgen action, and differences of gonadal and phenotypic sex.
5.1 Genetic Causes of 46,XY DSDs¶
- Disruption of Testis Development: WT1, SF1/NR5A1, SRY, SOX9, DHX37, DMRT1, CBX2, MAP3K1, SOX8, ZNRF3, GATA4/ZFPM2, DHH, ARX, TSPYL1, MYRF, ESR2/NR3A2, SAMD9, ATRX, PPP1R12A, MAMLD1.
- Disruption of Androgen Synthesis: LHCGR, DHCR7, STAR, CYP11A1, HSD3B2, CYP17A1, CYB5A, POR, HSD17B3, SRD5A2, AKR1C2.
- Disruption of Androgen Action: Androgen receptor.
Table 3 — Table 402-4 Selected Genetic Causes of 46,XY Differences of Sex Development (DSDs)¶
| GENE | INHERITANCE | GONAD | UTERUS | EXTERNAL GENITALIA | ASSOCIATED FEATURES |
|---|---|---|---|---|---|
| WT1 | AD | Dysgenetic testis | +/- | Female or ambiguous | Wilms’ tumor, renal abnormalities, gonadal tumors (WAGR, Denys-Drash and Frasier syndromes) |
| SF1/NR5A1 | AR/AD (SL) | Dysgenetic testis/Leydig dysfunction | +/- | Female, ambiguous or male | Primary adrenal failure (rare); hyposplenia (common); primary ovarian insufficiency in female (46,XX) relatives |
| SRY | Y | Dysgenetic testis or ovotestis | +/- | Female or ambiguous | |
| SOX9 | AD | Dysgenetic testis or ovotestis | +/- | Female or ambiguous | Campomelic dysplasia |
| DHX37 | AD | Dysgenetic testis | +/- | Female, ambiguous or male | Testicular regression syndrome |
| LHCGR | AR | Testis | - | Female, ambiguous or micropenis | Leydig cell hypoplasia |
| DHCR7 | AR | Testis | - | Variable | Smith-Lemli-Opitz syndrome: coarse facies, second-third toe syndactyly, failure to thrive, developmental delay, cardiac and visceral abnormalities |
| STAR | AR | Testis | - | Female or ambiguous | Congenital lipoid adrenal hyperplasia (primary adrenal insufficiency) |
| CYP11A1 | AR | Testis | - | Female or ambiguous | Primary adrenal insufficiency |
| HSD3B2 | AR | Testis | - | Ambiguous | CAH, primary adrenal insufficiency ± salt loss, partial androgenization due to ↑ DHEA |
| CYP17A1 | AR | Testis | - | Female or ambiguous | CAH, hypertension due to ↑ corticosterone and 11-deoxycorticosterone, except in isolated 17,20-lyase deficiency |
| CYB5A | AR | Testis | - | Ambiguous | Apparent isolated 17,20-lyase deficiency; methemoglobinemia |
| POR | AR | Testis | - | Ambiguous or male | Mixed features of 21-hydroxylase deficiency and 17α-hydroxylase/17,20-lyase deficiency, sometimes associated with Antley-Bixler craniosynostosis |
| HSD17B3 | AR | Testis | - | Female or ambiguous | Partial androgenization at puberty, ↑ androstenedione-to-testosterone ratio |
| SRD5A2 | AR | Testis | - | Ambiguous or micropenis | Partial androgenization at puberty, ↑ testosterone-to-dihydrotestosterone ratio |
| AKR1C2 | AR | Testis | - | Female or ambiguous | Decreased fetal DHT production |
| Androgen receptor | X | Testis | - | Female, ambiguous, micropenis or normal male | Phenotypic spectrum from complete androgen insensitivity syndrome (female external genitalia) and partial androgen insensitivity (ambiguous) to normal male genitalia and infertility |
6. INVESTIGATIONS & DIAGNOSIS¶
- Karyotype is a useful starting investigation for diagnosis with basic biochemical profiling.
- High-throughput genetic testing can help in reaching a definitive diagnosis.
- Serum sex steroids, AMH/MIS, and inhibin B are low, and LH and FSH are elevated in complete gonadal dysgenesis.
- Plasma concentrations of FSH and luteinizing hormone (LH) are increased in most adults with 47,XXY, and plasma testosterone is decreased (50–75%), reflecting primary gonadal insufficiency.
- Estradiol is often increased, resulting in gynecomastia.
- Those with mosaic forms of KS have less severe clinical features, have larger testes, and sometimes achieve spontaneous fertility.
- Individuals with CGD typically identify as female.
- Individuals raised as males often have hypospadias surgery and removal of dysgenetic or streak gonads if the gonads cannot be brought down into the scrotum.
- Scrotal testes can be preserved but require regular examination for tumor development and sonography (and possibly biopsy) at the time of puberty.
- Testosterone supplementation may be required in puberty or adulthood if low testosterone is detected.
- Individuals found to have congenital renal and urinary tract malformations (30%) are at risk for urinary tract infections, hypertension, and nephrocalcinosis.
- Regular assessment of thyroid function, weight, dentition, hearing, speech, vision, and educational issues should be performed during childhood.
6.1 Diagnostic Algorithm¶
- Initial Assessment: Evaluate for atypical genitalia, absent puberty, or primary amenorrhea.
- Karyotype Analysis: Determine chromosomal complement (46,XX, 46,XY, mosaicism).
- Hormonal Profiling: Measure FSH, LH, Testosterone, Estradiol, AMH/MIS, Inhibin B.
- Genetic Testing: High-throughput sequencing for specific gene mutations (SRY, SOX9, CYP21A2, etc.).
- Imaging: Ultrasound or MRI of gonads and internal structures (uterus, ovaries, kidneys).
- Cardiac Evaluation: Echocardiogram for Turner syndrome (coarctation, bicuspid aortic valve).
- Tumor Surveillance: Regular examination for germ cell tumors in individuals with Y chromosome material.
7. MANAGEMENT & TREATMENT¶
- Growth, endocrine function, and bone mineralization should be monitored, especially from adolescence.
- Educational and psychological support is important for many individuals with KS.
- The management of girls and women with TS requires a multidisciplinary approach to address many potentially affected organ systems according to TS practice guidelines.
- Individuals require long-term monitoring by an experienced cardiologist to follow congenital heart defects (CHDs).
- Regular assessment of thyroid function, weight, dentition, hearing, speech, vision, and educational issues should be performed during childhood.
- Counseling about long-term growth and fertility issues should be provided.
- Patient support groups are active throughout the world and can play an invaluable role.
- Short stature is common, and untreated final height rarely exceeds 150 cm in nonmosaic 45,X TS.
- Recombinant growth hormone is used to increase adult height.
- Girls with evidence of puberty at an age-appropriate time (~11 years) are treated with low-dose estrogen therapy to induce breast and uterine development, support growth, and maintain bone mineralization.
- Doses of estrogen are increased gradually to allow development over a 2- to 4-year period.
- Progestins are added later to regulate withdrawal bleeds.
- A very small percentage of women with TS have had spontaneous pregnancy, whereas others have achieved successful pregnancy after ovum donation and in vitro fertilization.
- Androgen supplementation improves virilization, libido, energy, hypofibrinolysis, and bone mineralization in men with low testosterone levels but may occasionally worsen gynecomastia.
- Gynecomastia can be treated by surgical reduction if it causes concern.
- Fertility has been achieved by using in vitro fertilization in men with oligospermia or with intracytoplasmic sperm injection (ICSI) after retrieval of spermatozoa by testicular sperm extraction techniques.
- Long-term follow-up of women with TS includes careful surveillance of sex hormone replacement and reproductive function, bone mineralization, cardiac function and aortic root dimensions, blood pressure, weight and glucose tolerance, hepatic and lipid profiles, thyroid function, celiac disease screening, skin examination, and hearing.
- This service is provided by a dedicated TS clinic in some centers.
7.1 Hormone Replacement Therapy¶
- Estrogen Replacement: Used for breast and uterine development, support growth, and maintain bone mineralization in Turner syndrome.
- Testosterone Supplementation: Used in men with low testosterone levels to improve virilization, libido, energy, hypofibrinolysis, and bone mineralization.
- Growth Hormone: Recombinant growth hormone is used to increase adult height in Turner syndrome.
- Progestins: Added later to regulate withdrawal bleeds in Turner syndrome.
7.2 Surgical and Procedural Management¶
- Gonadectomy: Traditionally advised for Turner syndrome if Y chromosome material exists due to tumor risk. Increasingly, the decision weighs potential for gonadal function and the importance of autonomous decision-making.
- Genitoplasty: Controversial; whether genitoplasty or prophylactic gonadectomy in selected conditions should be performed for infants and young children prior to the age of consent.
- Hypospadias Surgery: Individuals raised as males often have hypospadias surgery.
- Removal of Dysgenetic Gonads: Removal of dysgenetic or streak gonads if the gonads cannot be brought down into the scrotum.
- Testicular Prostheses: Can be offered to individuals with OTDSD.
8. PROGNOSIS & COMPLICATIONS¶
- KS: Increased risk of breast cancer, cardiovascular disease, metabolic syndrome, osteoporosis, and autoimmune disorders.
- TS: Increased risk of germ cell tumors, congenital heart defects (30%), coarctation of the aorta (30%), aortic root dilation (5%), hypertension, osteoporosis, autoimmune disorders.
- Ovotesticular DSD: Risk of GCC is also elevated in OTDSD (~3%).
- 45,X/46,XY Mosaicism: Increased risk of germ cell cancer (GCC), up to 35% in intraabdominal gonads.
- AIS: They also have an increased risk of GCC, again raising the question of if and when to perform gonadectomy.
- Persistent müllerian duct syndrome: Presence of a uterus in an otherwise phenotypic male.
- Hypospadias: Occurs in ~1 in 250 males.
8.1 Long-term Monitoring¶
- Cardiac Function: Follow congenital heart defects (CHDs) in Turner syndrome.
- Bone Mineralization: Monitor bone density and estrogen/testosterone levels.
- Metabolic Health: Monitor glucose tolerance, lipid profiles, and weight.
- Thyroid Function: Regular assessment of thyroid function.
- Hearing: Regular assessment of hearing.
- Skin: Skin examination for nevi and keloid formation.
- Educational Issues: Monitor for visuospatial learning difficulties and speech delay.
9. SPECIAL CONSIDERATIONS¶
- Pregnancy: A very small percentage of women with TS have had spontaneous pregnancy, whereas others have achieved successful pregnancy after ovum donation and in vitro fertilization, but the risks of cardiac complications are high.
- Mosaicism: 45,X/46,XX mosaicism and structural abnormalities of the X chromosome.
- Immunocompromised: Not explicitly detailed in the provided text.
- Renal Impairment: Individuals found to have congenital renal and urinary tract malformations (30%) are at risk for urinary tract infections, hypertension, and nephrocalcinosis.
10. KEY PEARLS & CLINICAL TRAPS¶
- Y chromosome material in individuals with TS increases the risk for germ cell tumors.
- Salt-losing crisis is a life-threatening presentation associated with congenital adrenal hyperplasia (CAH).
- Karyotype is a useful starting investigation for diagnosis with basic biochemical profiling.
- High-throughput genetic testing can help in reaching a definitive diagnosis.
- The existence of Y chromosome material in individuals with TS increases the risk for germ cell tumors, and gonadectomy has traditionally been advised.
- Gender variance and gender dysphoria are more common among some individuals with DSD than in the general population, though not high.
- Support groups also have a valuable role to play for many patients and families.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 402-1 Sex development can be divided into three major components: chromosomal sex, gonadal sex, and phenotypic sex. AMH, anti-müllerian hormone also known as Müllerian-inhibiting substance, MIS; DHT, dihydrotestosterone; T, testosterone. — Figure 402-1: Sex development can be divided into three major components: chromosomal sex, gonadal sex, and phenotypic sex.
Figure 2¶
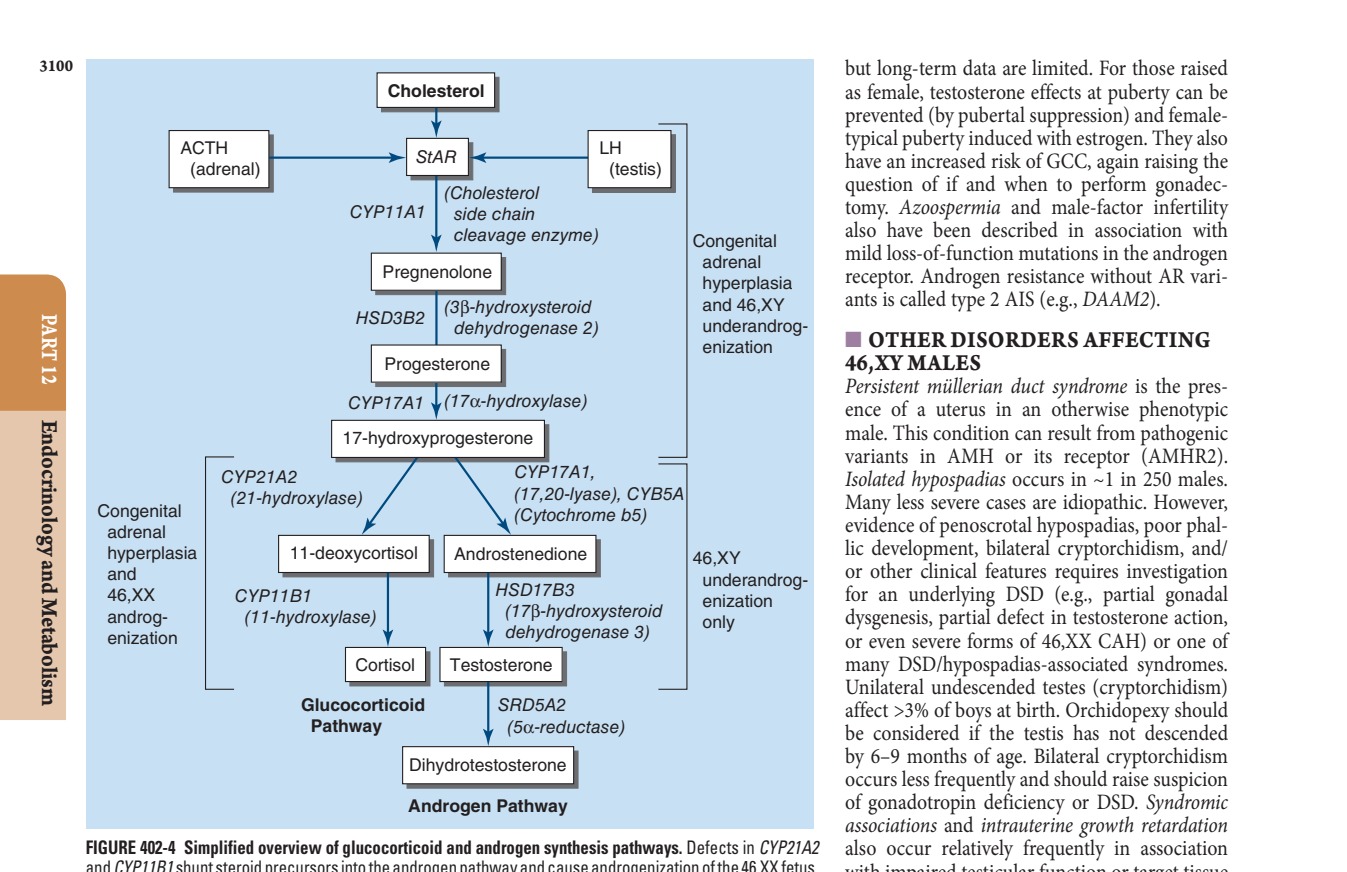
Caption: FIGURE 402-4 Simplified overview of glucocorticoid and androgen synthesis pathways. and CYP11B1 shunt steroid precursors into the androgen pathway and cause Testosterone is synthesized in the testicular Leydig cells and converted to Defects in enzymes involved in androgen synthesis result in underandrogenization of adrenocorticotropic hormone; LH, luteinizing hormone; StAR, steroidogenic acute — Figure 402-2: The genetic regulation of gonadal development showing the pathway from bipotential gonad to testis or ovary.
Figure 3¶
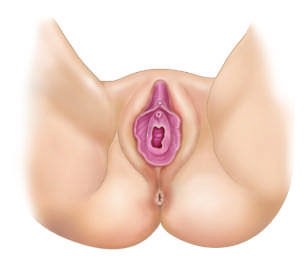
Caption: FIGURE 402-2 The genetic regulation of gonadal development. See text for additional genes involved. AMH, anti-müllerian hormone (müllerian-inhibiting substance); DHT, dihydrotestosterone; FOXL2, forkhead transcription factor L2; RSPO1, R-spondin 1; SF1, steroidogenic factor 1 (also known as NR5A1); SOX9, SRY- related HMG-box gene 9; SRY, sex-determining region on the Y chromosome; WNT4, wingless-type MMTV integration site 4; WT1, Wilms’ tumor–related gene 1. — Figure 402-3A: Internal urogenital tract showing the differentiation of Wolffian and Müllerian ducts into male or female structures.
Figure 4¶
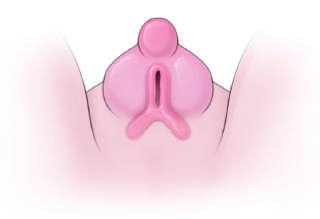
Caption: FIGURE 402-2 The genetic regulation of gonadal development. See text for additional genes involved. AMH, anti-müllerian hormone (müllerian-inhibiting substance); DHT, dihydrotestosterone; FOXL2, forkhead transcription factor L2; RSPO1, R-spondin 1; SF1, steroidogenic factor 1 (also known as NR5A1); SOX9, SRY- related HMG-box gene 9; SRY, sex-determining region on the Y chromosome; WNT4, wingless-type MMTV integration site 4; WT1, Wilms’ tumor–related gene 1. — Figure 402-3B: External genitalia showing the differentiation of the genital tubercle, urogenital swellings, and urethral folds.
Figure 5¶
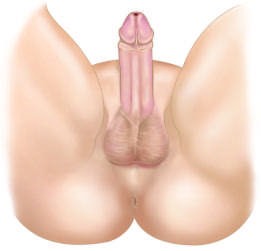
Caption: FIGURE 402-2 The genetic regulation of gonadal development. See text for additional genes involved. AMH, anti-müllerian hormone (müllerian-inhibiting substance); DHT, dihydrotestosterone; FOXL2, forkhead transcription factor L2; RSPO1, R-spondin 1; SF1, steroidogenic factor 1 (also known as NR5A1); SOX9, SRY- related HMG-box gene 9; SRY, sex-determining region on the Y chromosome; WNT4, wingless-type MMTV integration site 4; WT1, Wilms’ tumor–related gene 1. — Figure 402-4: Simplified overview of glucocorticoid and androgen synthesis pathways including enzymes like CYP21A2, CYP17A1, and SRD5A2.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.