Neuromyelitis Optica¶
Chapter 456 | Part 13: Neurologic Disorders · Part 13 – Neurologic Disorders
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- NMO is an aggressive, antibody-mediated inflammatory disorder characterized by recurrent attacks of optic neuritis (ON) and myelitis.
- The term NMOSD (Neuromyelitis Optica Spectrum Disorder) incorporates partial forms and additional CNS involvement.
- NMO is associated with anti-AQP4 antibodies in ~90% of patients; AQP4 is localized to astrocyte foot processes.
- Hallmark MRI finding: Longitudinally extensive transverse myelitis (LETM) extending over ≥3 contiguous vertebral segments.
- Area postrema syndrome (hiccups/vomiting) is a specific clinical feature of NMO.
- MS therapies (interferon beta, glatiramer acetate) are ineffective or harmful in NMO.
- MOGAD (MOG antibody-associated disease) presents with papillitis and fluffy brain lesions, distinct from NMO.
- GFAP autoimmunity presents with meningismus, encephalitis, and optic neuritis, often paraneoplastic.
- 5-year survival increased from 68–75% (1999) to 91–98% (2017) due to immune-suppressing therapies.
- AQP4-seronegative patients have lower relapse risk (~50% have single attack) but still require monitoring.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 NMOSD Classification
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 MOG Antibody-Associated Disease (MOGAD)
- 3.2 GFAP Autoimmunity
- 4. CLINICAL FEATURES
- 4.1 Core Clinical Characteristics
- 4.2 Associated Conditions
- 5. DIFFERENTIAL DIAGNOSIS
- 5.1 MS vs NMO Distinctions
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Diagnostic Criteria
- 6.2 Diagnostic Algorithm
- 7. MANAGEMENT & TREATMENT
- 7.1 Drug Therapy Details
- 7.2 Monitoring Parameters
- 8. PROGNOSIS & COMPLICATIONS
- 8.1 Survival & Disability
- 9. SPECIAL CONSIDERATIONS
- 9.1 Pediatric & Infectious Associations
- 10. KEY PEARLS & CLINICAL TRAPS
- Figures & Illustrations
📋 Figures in This Chapter¶
1. DEFINITION & OVERVIEW¶
📖 Harrison's defines this as:
NMO is an autoimmune disease associated with a highly specific autoantibody directed against the water channel protein AQP4 that is present in the sera of ~90% of affected patients.
- Neuromyelitis optica (NMO) is an aggressive, antibody-mediated, inflammatory disorder characterized by recurrent attacks of optic neuritis (ON) and myelitis.
- The more inclusive term Neuromyelitis Optica Spectrum Disorder (NMOSD) was proposed to incorporate individuals with partial forms and those with involvement of additional structures in the central nervous system.
- NMO is an autoimmune disease associated with a highly specific autoantibody directed against the water channel protein AQP4 that is present in the sera of ~90% of affected patients.
- AQP4 is localized to the foot processes of astrocytes in close apposition to endothelial surfaces, as well as at paranodal regions near nodes of Ranvier.
- Pathology of NMO shows loss of AQP4 associated with antibody and complement deposition and cytolysis of astrocytes within NMO.
- It is likely that AQP4 antibodies are pathogenic because passive transfer of AQP4 antibodies into laboratory animals can reproduce histologic features of the disease.
- Both complement fixation and antibody-dependent cell-mediated cytotoxicity (ADCC) are thought to contribute to astrocyte injury.
- Proinflammatory T lymphocytes of the T 17 type recognize an immunodominant epitope of AQP4 and may also contribute to pathogenesis.
- Recent data have identified a population of B lymphocytes in the thymus that constitutively express AQP4 and interact with AQP4-specific T cells to produce antigen-specific tolerance; this interaction might be impaired in NMO.
- During acute attacks of myelitis, CSF levels of interleukin-6 (IL-6; a proinflammatory cytokine) and glial fibrillary acidic protein (GFAP; an astrocyte protein) are markedly elevated, consistent with active inflammation and astrocyte injury.
- Because of the high specificity of the antibody, its presence is diagnostic when found in conjunction with a typical clinical presentation.
- Anti-AQP4 seropositive patients have a high risk for future attacks; more than half will relapse within 1 year if untreated.
1.1 NMOSD Classification¶
- NMOSD was proposed to incorporate individuals with partial forms and those with involvement of additional structures in the central nervous system.
- Table 456-1 outlines Diagnostic Criteria for NMOSD with AQP4-IgG and without AQP4-IgG or Unknown AQP4-IgG Status.
- Core clinical characteristics include optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome, and symptomatic cerebral syndrome with NMOSD-typical brain lesions.
2. EPIDEMIOLOGY¶
- NMO is more frequent in women than men (9:1).
- NMO typically begins in adulthood (40-year mean age of onset) but can arise at any age.
- The incidence and prevalence of NMO shows considerable variation between populations and geographic regions.
- Prevalence estimates range from 4 per 100,000.
- Although NMO can occur in people of any ethnic background, individuals of Asian and African origin are disproportionately affected.
- The highest reported prevalence is in Martinique.
- Among white populations, MS (Chap. 455) is far more common than NMO.
- In some NMO cases, onset may be associated with acute infection that with varicella-zoster virus, Epstein-Barr virus, HIV, or tuberculosis.
- Rare cases appear to be paraneoplastic and associated with breast, lung, or other cancers.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- NMO is an autoimmune disease associated with a highly specific autoantibody directed against the water channel protein AQP4.
- AQP4 is localized to the foot processes of astrocytes in close apposition to endothelial surfaces, as well as at paranodal regions near nodes of Ranvier.
- Pathology of NMO shows loss of AQP4 associated with antibody and complement deposition and cytolysis of astrocytes within NMO.
- It is likely that AQP4 antibodies are pathogenic because passive transfer of AQP4 antibodies into laboratory animals can reproduce histologic features of the disease.
- Both complement fixation and antibody-dependent cell-mediated cytotoxicity (ADCC) are thought to contribute to astrocyte injury.
- Proinflammatory T lymphocytes of the T 17 type recognize an immunodominant epitope of AQP4 and may also contribute to pathogenesis.
- Recent data have identified a population of B lymphocytes in the thymus that constitutively express AQP4 and interact with AQP4-specific T cells to produce antigen-specific tolerance; this interaction might be impaired in NMO.
- During acute attacks of myelitis, CSF levels of interleukin-6 (IL-6; a proinflammatory cytokine) and glial fibrillary acidic protein (GFAP; an astrocyte protein) are markedly elevated, consistent with active inflammation and astrocyte injury.
- Up to 40% of NMO patients have a systemic autoimmune disorder, such as systemic lupus erythematosus, Sjögren's syndrome, perinuclear antineutrophil cytoplasmic antibody (p-ANCA)–associated vasculitis, myasthenia gravis, Hashimoto's thyroiditis, or mixed connective tissue disease.
- This is another feature distinct from MS; MS patients rarely have other comorbid autoimmune diseases other than hypothyroidism.
- In some NMO cases, onset may be associated with acute infection that with varicella-zoster virus, Epstein-Barr virus, HIV, or tuberculosis.
- Rare cases appear to be paraneoplastic and associated with breast, lung, or other cancers.
3.1 MOG Antibody-Associated Disease (MOGAD)¶
- Anti–myelin oligodendrocyte glycoprotein (MOG) antibodies detected by a cell-based assay that enables recognition of MOG epitopes in a lipid bilayer were only recently found to be associated with cases of acute disseminated encephalomyelitis (ADEM) in human children and then with cases of AQP4-seronegative NMO.
- Patients who are seropositive for anti-MOG antibodies are at risk for bilateral, synchronous optic neuritis and myelitis and meningoencephalitis.
- A clinical feature that can distinguish ON associated with MOG antibody–associated disease (MOGAD) from NMO or MS is the presence of papillitis seen by fundoscopy or orbital MRI, a finding that is common in MOGAD, rare in NMO, and variable but usually unilateral in MS.
- ON associated with MOGAD is typically longitudinally extensive on MRI, and brain MRI can be normal or show fluffy areas of increased signal change in white or gray matter structures, similar to NMO.
- MRI lesions that are typical for MS, including finger-like lesions oriented perpendicular to the ventricular surface (Dawson fingers) and T1 hypointense lesions, are uncommon.
- Spinal cord lesions can be longitudinally extensive or short and sometimes involve the conus medullaris.
- Demyelination associated with MOGAD is sometimes monophasic, as in ADEM, but can also be recurrent.
- The CSF may show a pleocytosis with occasional neutrophils.
- Elevated intrathecal synthesis of gamma globulins is atypical: oligoclonal bands are present in only ~6–13% of cases.
- A small percentage of patients who present with syndromes suggestive of MOGAD but test negative (or only weakly positive) for serum anti-MOG antibodies have detectable antibodies restricted to the CSF; thus, in these seronegative cases, CSF anti-MOG antibody testing should be pursued.
- The mechanism of central nervous system (CNS) injury in MOGAD is not established and could involve MOG-reactive cytotoxic T cells in addition to anti-MOG antibodies.
- Studies in MOG-induced experimental autoimmune encephalomyelitis, an animal model, suggest that anti-MOG antibodies may opsonize traces of MOG protein in secondary lymphoid tissues, triggering a peripheral immune response against MOG.
- Brain lesions associated with MOGAD often respond rapidly to treatment with glucocorticoids and may resolve entirely.
- Some patients experience disease recurrence following discontinuation of prednisone and can become glucocorticoid dependent.
- Clinical trials in MOGAD are underway with satralizumab (the IL-6 receptor blocker indicated for AQP4-seropositive NMO) and rozanolixizumab (Rystiggo; a humanized monoclonal antibody that binds the neonatal Fc receptor).
- There are limited data on use of other immune-suppressing medications typically used in NMO.
- Off-label empiric treatments include daily prednisone, IV immunoglobulin, rituximab, and mycophenolate mofetil.
- Anti-MOG antibody titers appear to decline either spontaneously or in the setting of treatment.
3.2 GFAP Autoimmunity¶
- Autoimmunity against the astrocyte protein GFAP presents with a range of symptoms referable to meningismus, encephalitis, myelitis, and optic neuritis.
- Some cases follow a viral prodrome, and fever and headache are commonly present.
- The clinical syndrome can also include a movement disorder reflecting involvement of deep gray matter structures.
- MRI shows characteristic patterns of gadolinium enhancement localized to GFAP-enriched CNS regions including venous structures in a periventricular radial orientation, the leptomeninges, the periependymal spinal cord, and a striking serpiginous pattern involving brain parenchyma.
- Longitudinally extensive spinal cord involvement can also be present.
- These patterns share similarities with those observed in neurosarcoidosis, and their presence should prompt consideration for either condition.
- A lymphocytic pleocytosis is commonly present in the CSF.
- Antibodies against GFAP can be measured in the serum or CSF.
- GFAP autoimmunity is found as a paraneoplastic syndrome in ~25% of cases, most commonly associated with ovarian teratoma, and can coexist with anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis or AQP4 NMO.
- T cells are implicated in pathophysiology based on histopathology and association with checkpoint inhibitor treatment for cancer or in the setting of HIV.
- GFAP autoimmunity is generally glucocorticoid responsive.
- Early recognition with prompt intervention is associated with more favorable outcomes.
- Relapses occur in ~20% of patients and require use of immune suppression therapy.
4. CLINICAL FEATURES¶
- NMO is typically a recurrent disease; the course is monophasic in <10% of patients.
- Individuals who test negative for AQP4 antibodies are somewhat more likely to have a monophasic course.
- Untreated NMO is usually severely disabling over time; in one series, respiratory failure from cervical myelitis was present in one-third of patients.
- 8 years after onset, 60% of patients were blind and more than half had permanent paralysis of one or more limbs.
- Observational data suggest that the long-term course of NMO has been substantially improved since the development of therapies to treat acute attacks and prevent relapses.
- The 5-year survival rate appears to have increased from 68–75% around 1999 to 91–98% as of 2017, a change presumably due to improved diagnosis and widespread use of immune-suppressing therapies.
- In patients with NMO, attacks of ON can be bilateral and produce severe vision loss (uncommon in MS).
- Myelitis and an accumulation of disability is caused by attack-related injury.
- Progressive symptoms only rarely occur in NMO.
- Brain MRI lesions are present, including areas of nonspecific signal change as well as lesions associated with specific syndromes such as the area postrema in the lower medulla presenting as intractable hiccoughs or vomiting.
- Lesions in the hypothalamus causing a sleep disorder or endocrinopathy.
- Lesions in the cerebral hemispheres producing focal symptoms, encephalopathy, or seizures.
- Large MRI lesions in the cerebral hemispheres can be asymptomatic, sometimes have a 'cloud-like' appearance, and unlike MS lesions, are often not destructive and can resolve completely.
- Spinal cord MRI lesions typically consist of focal enhancing areas of swelling and tissue destruction, extending over three or more spinal cord segments, and on axial sequences, these are centered on the gray matter of the cord.
- Cerebrospinal fluid (CSF) findings include pleocytosis greater than that observed in MS, with neutrophils and eosinophils present in many acute cases.
- Oligoclonal bands (OCBs) are uncommon, occurring in <20% of NMO patients.
4.1 Core Clinical Characteristics¶
-
- Optic neuritis.
-
- Acute myelitis.
-
- Area postrema syndrome: episode of otherwise unexplained hiccups or nausea or vomiting.
-
- Acute brainstem syndrome.
-
- Symptomatic narcolepsy or acute diencephalic clinical syndrome (hypothalamic dysfunction) with NMOSD-typical diencephalic MRI lesions.
-
- Symptomatic cerebral syndrome with NMOSD-typical brain lesions.
4.2 Associated Conditions¶
- Up to 40% of NMO patients have a systemic autoimmune disorder.
- Examples include systemic lupus erythematosus, Sjögren's syndrome, perinuclear antineutrophil cytoplasmic antibody (p-ANCA)–associated vasculitis, myasthenia gravis, Hashimoto's thyroiditis, or mixed connective tissue disease.
- This is another feature distinct from MS; MS patients rarely have other comorbid autoimmune diseases other than hypothyroidism.
5. DIFFERENTIAL DIAGNOSIS¶
- Distinguishing between NMO and multiple sclerosis (MS) is an important consideration, especially early in its presentation.
- In patients with NMO, attacks of ON can be bilateral and produce severe vision loss (uncommon in MS).
- Myelitis in NMO is typically longitudinally extensive (rare in MS) and is typically longitudinally extensive involving three or more contiguous vertebral segments.
- In contrast to MS, progressive symptoms only rarely occur in NMO, and accumulation of disability is caused by attack-related injury.
- MS lesions are typically periventricular and oriented perpendicular to the ventricular surface (Dawson fingers).
- T1 hypointense lesions are common in MS but uncommon in NMO.
- MOGAD presents with papillitis (common in MOGAD, rare in NMO, variable in MS).
- ADEM has a monophasic course and is often associated with an antecedent infection (postinfectious encephalomyelitis).
- ADEM is far more common in children than adults, and many adult cases initially thought to represent ADEM subsequently experience late relapses qualifying as either MS or NMOSD.
- GFAP autoimmunity presents with meningismus, encephalitis, myelitis, and optic neuritis, often paraneoplastic.
5.1 MS vs NMO Distinctions¶
- MS lesions are typically periventricular and oriented perpendicular to the ventricular surface (Dawson fingers).
- T1 hypointense lesions are common in MS but uncommon in NMO.
- Spinal cord lesions in MS are typically short and sometimes involve the conus medullaris.
- Demyelination associated with MOGAD is sometimes monophasic, as in ADEM, but can also be recurrent.
- The CSF may show a pleocytosis with occasional neutrophils in MOGAD.
- Elevated intrathecal synthesis of gamma globulins is atypical in MOGAD: oligoclonal bands are present in only ~6–13% of cases.
6. INVESTIGATIONS & DIAGNOSIS¶
- Brain magnetic resonance imaging (MRI) was earlier thought to be normal in NMO, but it is now recognized that in many cases brain lesions are present, including areas of nonspecific signal change as well as lesions associated with specific syndromes.
- Spinal cord MRI lesions typically consist of focal enhancing areas of swelling and tissue destruction, extending over three or more spinal cord segments, and on axial sequences, these are centered on the gray matter of the cord.
- Cerebrospinal fluid (CSF) findings include pleocytosis greater than that observed in MS, with neutrophils and eosinophils present in many acute cases.
- Oligoclonal bands (OCBs) are uncommon, occurring in 1/2 optic nerve length or involving optic chiasm.
- Acute myelitis requires associated intramedullary MRI lesion extending ≥3 contiguous segments (LETM) OR ≥3 contiguous segments of focal spinal cord atrophy in patients with history compatible with acute myelitis.
- Area postrema syndrome requires associated dorsal medulla/area postrema lesions.
- Acute brainstem syndrome requires periependymal brainstem lesions.
6.1 Diagnostic Criteria¶
- Diagnostic Criteria for NMOSD with AQP4-IgG: 1. At least one core clinical characteristic. 2. Positive test for AQP4-IgG using best available detection method (cell-based assay strongly recommended). 3. Exclusion of alternative diagnoses.
- Diagnostic Criteria for NMOSD without AQP4-IgG or NMOSD with Unknown AQP4-IgG Status: 1. At least two core clinical characteristics occurring as a result of one or more clinical attacks and meeting all of the following requirements: a. At least one core clinical characteristic must be optic neuritis, acute myelitis with LETM, or area postrema syndrome. b. Dissemination in space (two or more different clinical characteristics). c. Fulfillment of additional MRI requirements, as applicable. 2. Negative test for AQP4-IgG using best available detection method or testing unavailable. 3. Exclusion of alternative diagnoses.
- Core Clinical Characteristics: 1. Optic neuritis. 2. Acute myelitis. 3. Area postrema syndrome: episode of otherwise unexplained hiccups or nausea or vomiting. 4. Acute brainstem syndrome. 5. Symptomatic narcolepsy or acute diencephalic clinical syndrome (hypothalamic dysfunction) with NMOSD-typical diencephalic MRI lesions. 6. Symptomatic cerebral syndrome with NMOSD-typical brain lesions.
- Additional MRI Requirements for NMOSD without AQP4-IgG and NMOSD with Unknown AQP4-IgG Status: 1. Acute optic neuritis: requires brain MRI showing (a) normal findings or only nonspecific white matter lesions, OR (b) optic nerve MRI with T2-hyperintense lesion of T1-weighted gadolinium-enhancing lesion extending over >1/2 optic nerve length or involving optic chiasm. 2. Acute myelitis: requires associated intramedullary MRI lesion extending ≥3 contiguous segments (LETM) OR ≥3 contiguous segments of focal spinal cord atrophy in patients with history compatible with acute myelitis. 3. Area postrema syndrome requires associated dorsal medulla/area postrema lesions. 4. Acute brainstem syndrome requires periependymal brainstem lesions.
6.2 Diagnostic Algorithm¶
- Step 1: Assess clinical presentation for core characteristics (optic neuritis, acute myelitis, area postrema syndrome, etc.).
- Step 2: Perform MRI of brain and spinal cord to evaluate for LETM, optic nerve lesions, and area postrema lesions.
- Step 3: Test for AQP4-IgG using cell-based assay.
- Step 4: If AQP4-IgG positive, diagnosis of NMOSD is established (with exclusion of alternative diagnoses).
- Step 5: If AQP4-IgG negative or unknown, test for MOG-IgG.
- Step 6: If MOG-IgG positive, evaluate for MOGAD criteria (core clinical event, supporting features, exclusion of MS).
- Step 7: If MOG-IgG negative, test for GFAP antibodies.
- Step 8: If GFAP antibodies positive, evaluate for GFAP autoimmunity criteria.
- Step 9: If all antibody tests negative, consider alternative diagnoses (MS, ADEM, infections, etc.).
7. MANAGEMENT & TREATMENT¶
- Acute attacks are usually treated with high-dose glucocorticoids (e.g., methylprednisolone 1 g/d for 5–10 days followed by a prednisone taper).
- Plasma exchange (typically 5–7 exchanges of 1.5 plasma volumes/exchange) is used empirically for acute episodes that do not respond to glucocorticoids.
- Given the unfavorable natural history of untreated NMO, prophylaxis against relapses is recommended for nearly all patients.
- Four monoclonal antibody medications are approved for attack prevention in NMO: an anti-CD19 B-cell depleter (inebilizumab), an IL-6 receptor blocker (satralizumab), and two terminal complement inhibitors (eculizumab and ravulizumab).
- It is our general practice to begin therapy with either inebilizumab or satralizumab and to use complement inhibitors as second-line treatment for nonresponders.
- None of the U.S. Food and Drug Administration–approved medications are approved for AQP4-negative patients, and in practice, obtaining these medications for seronegative patients is challenging from a payer perspective.
- In AQP4-seronegative patients, the risk of a relapse is lower (approximately half of patients have only a single attack), and in these patients, our practice is to begin treatment with empiric therapies such as rituximab or mycophenolate mofetil.
- Because AQP4-seronegative patients require involvement of both optic neuritis and longitudinally extensive myelitis to meet criteria for seronegative NMOSD, the majority of AQP4-seronegative patients will have had more than one attack.
- Inebilizumab (Uplizna): Inebilizumab is a humanized affinity-optimized, afucosylated monoclonal antibody that binds to the B-cell surface antigen CD19 and depletes a wide range of B cells including pre-B cells and some plasmablasts.
- In the pivotal clinical trial that included individuals both positive and negative for AQP4, inebilizumab reduced the time to the first attack by 77% compared to placebo (hazard ratio, 0.23; p <.0001).
- Inebilizumab-treated participants also had reduced rates of hospitalizations, disability worsening, and new MRI lesions.
- Inebilizumab is dosed as an initial 300-mg IV infusion followed 2 weeks later by a second 300-mg IV infusion, with subsequent doses of 300-mg infusions every 6 months thereafter.
- Inebilizumab is associated with a dose dependent decline in serum IgG levels and with neutropenia in some patients.
- Satralizumab (Enspryng): Satralizumab is a monoclonal antibody that binds to the IL-6 receptor, blocking engagement of IL-6, a proinflammatory cytokine that is upregulated in the CSF during acute NMO attacks.
- Satralizumab was investigated in NMOSD in two registration trials: one as monotherapy and the other as add-on therapy.
- Both AQP4-seropositive and AQP4-seronegative participants were enrolled.
- In the monotherapy study, the risk of attack was reduced by 74% with satralizumab (p = .014), and in the add-on study, the risk of attack was reduced by 78% (p = .014).
- Although both studies recruited substantial numbers of AQP4-seronegative participants, there was no clinically meaningful impact of satralizumab in seronegative participants.
- Satralizumab is administered as a loading dosage of 120 mg by subcutaneous injection at weeks 0, 2, and 4, followed by a maintenance dose of 120 mg every 4 weeks.
- Screening for hepatitis B virus, tuberculosis, and liver transaminase elevations is required before starting satralizumab.
- Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) should be monitored during treatment for transaminase elevations and complete blood count (CBC) monitored for neutropenia.
- Satralizumab is also associated with weight gain; body weight increases of at least 7% from baseline occurred in 30% of satralizumab-treated participants compared to 8% of those treated with placebo.
- Eculizumab (Soliris): The monoclonal antibody eculizumab binds with high affinity to the complement protein C5, inhibiting its cleavage into C5a and C5b and thereby preventing generation of the terminal complement attack complex C5b-9.
- Eculizumab was investigated as an add-on therapy in AQP4-seropositive NMO.
- The time to first attack was longer in patients treated with eculizumab compared to placebo (relative risk reduction, 94%; hazard ratio, 0.058; p <.0001).
- Eculizumab-treated patients also had a 96% relative reduction in the annualized attack rate, as well as dramatically reduced rates of hospitalizations and need for glucocorticoids or plasma exchange for acute attacks.
- Eculizumab is dosed as follows: 900 mg weekly for the first 4 weeks, followed by 1200 mg for the fifth dose 1 week later, and then 1200 mg every 2 weeks thereafter.
- Because of its toxicity profile, eculizumab is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS).
- Life-threatening and fatal meningococcal infections have occurred (boxed warning).
- Eculizumab-treated patients must be immunized with meningococcal vaccines (MenACWY two doses at least 8 weeks apart plus either MenB-4C [two doses at least 1 month apart] or MenB-FHbp [three doses over 6 months]) at least 2 weeks prior to administering the first dose unless the risks of delaying eculizumab therapy outweigh the risks of developing a meningococcal infection.
- The MenACWY vaccine requires revaccination every 5 years, and the MenB-4C/MenB-FHbp requires a booster 1 year after vaccination and then every 2–3 years with ongoing treatment.
- Vaccination reduces, but does not eliminate, the risk of meningococcal infections.
- Eculizumab-treated patients must be monitored closely for early signs of meningococcal infections and evaluated immediately if infection is suspected.
- Ravulizumab (Ultomiris): Like eculizumab, ravulizumab is a terminal complement inhibitor.
- Ravulizumab was developed using antibody recycling technology to increase its half-life, thereby allowing less frequent administration than its parent compound eculizumab.
- In a single-arm, open-label study of ravulizumab in NMOSD, ravulizumab-treated participants did not experience any attacks during 50 weeks of observation.
- A historical control arm from the eculizumab PREVENT clinical trial was used for comparison.
- Ravulizumab-treated patients also require vaccination for meningococcal infections.
- It seems likely that ravulizumab produces even more potent complement inhibition than eculizumab, resulting in apparently greater efficacy but also a higher risk of meningococcal infection.
- Ravulizumab is administered intravenously with a weight-based loading dose followed by maintenance dosing every 8 weeks.
- Prior to the approval of disease-modifying therapies for NMO, several empiric regimens were commonly used including mycophenolate mofetil (1000 mg bid); rituximab, a B-cell-depleting anti-CD20 monoclonal antibody (2 g IV every 6 months); or a combination of glucocorticoids (500 mg IV methylprednisolone daily for 5 days, then oral prednisone 1 mg/kg per day for 2 months, followed by slow taper) plus azathioprine (2 mg/kg per day started on week 3).
- Plasma cell–targeted therapy with anti–B-cell maturation antigen (BCMA) chimeric antigen receptor (CAR) T cells has shown promise in an early clinical study of patients with NMOSD who failed to respond to other treatments.
- Importantly, some therapies with proven efficacy in MS do not appear to be useful for NMO.
- Available evidence suggests that interferon beta is ineffective and paradoxically may increase the risk of NMO relapses, and based on limited data, glatiramer acetate, fingolimod, natalizumab, and alemtuzumab also appear to be ineffective.
- These distinctions highlight the need for efficient diagnosis of NMO.
- Acute episodes are managed with high-dose glucocorticoids followed by a prednisone taper and sometimes by plasmapheresis, as with NMO.
7.1 Drug Therapy Details¶
- Inebilizumab: Dose 300-mg IV infusion initial, 300-mg IV infusion 2 weeks later, subsequent doses 300-mg infusions every 6 months thereafter. Side effects: Dose dependent decline in serum IgG levels, neutropenia.
- Satralizumab: Dose 120 mg subcutaneous injection weeks 0, 2, 4 (loading), then 120 mg every 4 weeks (maintenance). Side effects: Weight gain (30% increase ≥7%), neutropenia, hepatitis B, tuberculosis, liver transaminase elevations.
- Eculizumab: Dose 900 mg weekly for first 4 weeks, 1200 mg for fifth dose 1 week later, then 1200 mg every 2 weeks thereafter. Side effects: Meningococcal infections (boxed warning), requires meningococcal vaccination.
- Ravulizumab: Dose weight-based loading dose followed by maintenance dosing every 8 weeks. Side effects: Meningococcal infections (requires vaccination).
- Rituximab: Dose 2 g IV every 6 months. Side effects: Infusion reactions, infections.
- Mycophenolate mofetil: Dose 1000 mg bid. Side effects: GI upset, leukopenia.
7.2 Monitoring Parameters¶
- Screening for hepatitis B virus, tuberculosis, and liver transaminase elevations is required before starting satralizumab.
- Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) should be monitored during treatment for transaminase elevations.
- Complete blood count (CBC) monitored for neutropenia.
- Eculizumab-treated patients must be monitored closely for early signs of meningococcal infections and evaluated immediately if infection is suspected.
- Inebilizumab is associated with a dose dependent decline in serum IgG levels and with neutropenia in some patients.
8. PROGNOSIS & COMPLICATIONS¶
- Untreated NMO is usually severely disabling over time.
- In one series, respiratory failure from cervical myelitis was present in one-third of patients.
- 8 years after onset, 60% of patients were blind and more than half had permanent paralysis of one or more limbs.
- The 5-year survival rate appears to have increased from 68–75% around 1999 to 91–98% as of 2017, a change presumably due to improved diagnosis and widespread use of immune-suppressing therapies.
- Anti-AQP4 seropositive patients have a high risk for future attacks; more than half will relapse within 1 year if untreated.
- Individuals who test negative for AQP4 antibodies are somewhat more likely to have a monophasic course.
- Relapses occur in ~20% of patients with GFAP autoimmunity and require use of immune suppression therapy.
- Some patients experience disease recurrence following discontinuation of prednisone and can become glucocorticoid dependent (MOGAD).
- Clinical trials in MOGAD are underway with satralizumab and rozanolizumab.
8.1 Survival & Disability¶
- 5-year survival rate increased from 68–75% (1999) to 91–98% (2017).
- 8 years after onset, 60% of patients were blind and more than half had permanent paralysis of one or more limbs.
- Respiratory failure from cervical myelitis was present in one-third of patients in one series.
9. SPECIAL CONSIDERATIONS¶
- In some NMO cases, onset may be associated with acute infection that with varicella-zoster virus, Epstein-Barr virus, HIV, or tuberculosis.
- Rare cases appear to be paraneoplastic and associated with breast, lung, or other cancers.
- GFAP autoimmunity is found as a paraneoplastic syndrome in ~25% of cases, most commonly associated with ovarian teratoma, and can coexist with anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis or AQP4 NMO.
- T cells are implicated in pathophysiology based on histopathology and association with checkpoint inhibitor treatment for cancer or in the setting of HIV.
- ADEM is far more common in children than adults, and many adult cases initially thought to represent ADEM subsequently experience late relapses qualifying as either MS or NMOSD.
- Clinical trials in MOGAD are underway with satralizumab (the IL-6 receptor blocker indicated for AQP4-seropositive NMO) and rozanolizumab (Rystiggo; a humanized monoclonal antibody that binds the neonatal Fc receptor).
- There are limited data on use of other immune-suppressing medications typically used in NMO.
- Off-label empiric treatments include daily prednisone, IV immunoglobulin, rituximab, and mycophenolate mofetil.
9.1 Pediatric & Infectious Associations¶
- ADEM is far more common in children than adults.
- Many adult cases initially thought to represent ADEM subsequently experience late relapses qualifying as either MS or NMOSD.
- Some cases follow a viral prodrome, and fever and headache are commonly present.
- In some NMO cases, onset may be associated with acute infection that with varicella-zoster virus, Epstein-Barr virus, HIV, or tuberculosis.
- Rare cases appear to be paraneoplastic and associated with breast, lung, or other cancers.
10. KEY PEARLS & CLINICAL TRAPS¶
- NMO is an aggressive, antibody-mediated, inflammatory disorder characterized by recurrent attacks of optic neuritis (ON) and myelitis.
- The more inclusive term NMOSD was proposed to incorporate individuals with partial forms and those with involvement of additional structures in the central nervous system.
- NMO is more frequent in women than men (9:1) and typically begins in adulthood (40-year mean age of onset) but can arise at any age.
- An important consideration, especially early in its presentation, is distinguishing between NMO and multiple sclerosis (MS).
- In patients with NMO, attacks of ON can be bilateral and produce severe vision loss (uncommon in MS).
- Myelitis and an accumulation of disability is caused by attack-related injury.
- Progressive symptoms only rarely occur in NMO.
- Large MRI lesions in the cerebral hemispheres can be asymptomatic, sometimes have a 'cloud-like' appearance, and unlike MS lesions, are often not destructive and can resolve completely.
- Spinal cord MRI lesions typically consist of focal enhancing areas of swelling and tissue destruction, extending over three or more spinal cord segments, and on axial sequences, these are centered on the gray matter of the cord.
- Cerebrospinal fluid (CSF) findings include pleocytosis greater than that observed in MS, with neutrophils and eosinophils present in many acute cases.
- Oligoclonal bands (OCBs) are uncommon, occurring in <20% of NMO patients.
- Up to 40% of NMO patients have a systemic autoimmune disorder, such as systemic lupus erythematosus, Sjögren's syndrome, perinuclear antineutrophil cytoplasmic antibody (p-ANCA)–associated vasculitis, myasthenia gravis, Hashimoto's thyroiditis, or mixed connective tissue disease.
- This is another feature distinct from MS; MS patients rarely have other comorbid autoimmune diseases other than hypothyroidism.
- Available evidence suggests that interferon beta is ineffective and paradoxically may increase the risk of NMO relapses, and based on limited data, glatiramer acetate, fingolimod, natalizumab, and alemtuzumab also appear to be ineffective.
- These distinctions highlight the need for efficient diagnosis of NMO.
- Acute episodes are managed with high-dose glucocorticoids followed by a prednisone taper and sometimes by plasmapheresis, as with NMO.
- Given the unfavorable natural history of untreated NMO, prophylaxis against relapses is recommended for nearly all patients.
- Four monoclonal antibody medications are approved for attack prevention in NMO: an anti-CD19 B-cell depleter (inebilizumab), an IL-6 receptor blocker (satralizumab), and two terminal complement inhibitors (eculizumab and ravulizumab).
- It is our general practice to begin therapy with either inebilizumab or satralizumab and to use complement inhibitors as second-line treatment for nonresponders.
- None of the U.S. Food and Drug Administration–approved medications are approved for AQP4-negative patients, and in practice, obtaining these medications for seronegative patients is challenging from a payer perspective.
- In AQP4-seronegative patients, the risk of a relapse is lower (approximately half of patients have only a single attack), and in these patients, our practice is to begin treatment with empiric therapies such as rituximab or mycophenolate mofetil.
- Because AQP4-seronegative patients require involvement of both optic neuritis and longitudinally extensive myelitis to meet criteria for seronegative NMOSD, the majority of AQP4-seronegative patients will have had more than one attack.
- A clinical feature that can distinguish ON associated with MOG antibody–associated disease (MOGAD) from NMO or MS is the presence of papillitis seen by fundoscopy or orbital MRI, a finding that is common in MOGAD, rare in NMO, and variable but usually unilateral in MS.
- ON associated with MOGAD is typically longitudinally extensive on MRI, and brain MRI can be normal or show fluffy areas of increased signal change in white or gray matter structures, similar to NMO.
- MRI lesions that are typical for MS, including finger-like lesions oriented perpendicular to the ventricular surface (Dawson fingers) and T1 hypointense lesions, are uncommon.
- Spinal cord lesions can be longitudinally extensive or short and sometimes involve the conus medullaris.
- Demyelination associated with MOGAD is sometimes monophasic, as in ADEM, but can also be recurrent.
- The CSF may show a pleocytosis with occasional neutrophils.
- Elevated intrathecal synthesis of gamma globulins is atypical: oligoclonal bands are present in only ~6–13% of cases.
- A small percentage of patients who present with syndromes suggestive of MOGAD but test negative (or only weakly positive) for serum anti-MOG antibodies have detectable antibodies restricted to the CSF; thus, in these seronegative cases, CSF anti-MOG antibody testing should be pursued.
- The mechanism of central nervous system (CNS) injury in MOGAD is not established and could involve MOG-reactive cytotoxic T cells in addition to anti-MOG antibodies.
- Studies in MOG-induced experimental autoimmune encephalomyelitis, an animal model, suggest that anti-MOG antibodies may opsonize traces of MOG protein in secondary lymphoid tissues, triggering a peripheral immune response against MOG.
- Brain lesions associated with MOGAD often respond rapidly to treatment with glucocorticoids and may resolve entirely.
- Some patients experience disease recurrence following discontinuation of prednisone and can become glucocorticoid dependent.
- Clinical trials in MOGAD are underway with satralizumab (the IL-6 receptor blocker indicated for AQP4-seropositive NMO) and rozanolizumab (Rystiggo; a humanized monoclonal antibody that binds the neonatal Fc receptor).
- There are limited data on use of other immune-suppressing medications typically used in NMO.
- Off-label empiric treatments include daily prednisone, IV immunoglobulin, rituximab, and mycophenolate mofetil.
- Anti-MOG antibody titers appear to decline either spontaneously or in the setting of treatment.
- Autoimmunity against the astrocyte protein GFAP presents with a range of symptoms referable to meningismus, encephalitis, myelitis, and optic neuritis.
- Some cases follow a viral prodrome, and fever and headache are commonly present.
- The clinical syndrome can also include a movement disorder reflecting involvement of deep gray matter structures.
- MRI shows characteristic patterns of gadolinium enhancement localized to GFAP-enriched CNS regions including venous structures in a periventricular radial orientation, the leptomeninges, the periependymal spinal cord, and a striking serpiginous pattern involving brain parenchyma.
- Longitudinally extensive spinal cord involvement can also be present.
- These patterns share similarities with those observed in neurosarcoidosis, and their presence should prompt consideration for either condition.
- A lymphocytic pleocytosis is commonly present in the CSF.
- Antibodies against GFAP can be measured in the serum or CSF.
- GFAP autoimmunity is found as a paraneoplastic syndrome in ~25% of cases, most commonly associated with ovarian teratoma, and can coexist with anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis or AQP4 NMO.
- T cells are implicated in pathophysiology based on histopathology and association with checkpoint inhibitor treatment for cancer or in the setting of HIV.
- GFAP autoimmunity is generally glucocorticoid responsive.
- Early recognition with prompt intervention is associated with more favorable outcomes.
- Relapses occur in ~20% of patients and require use of immune suppression therapy.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 456-1 Imaging findings in neuromyelitis optica: longitudinally extensive inversion recovery (FLAIR) cervical spine MRI showing an area of increased signal B. Sagittal T1-weighted cervical spine MRI following gadolinium-diethylene triamine brain MRI following gadolinium DPTA infusion shows enhancement of the right optic within the area postrema (arrow). F. Axial T1-weighted brain MRI following — Sagittal fluid-attenuated inversion recovery (FLAIR) cervical spine MRI showing an area of increased signal change on T2-weighted imaging spanning more than three vertebral segments in length, characteristic of longitudinally extensive transverse myelitis.
Figure 2¶

Caption: FIGURE 456-1 Imaging findings in neuromyelitis optica: longitudinally extensive inversion recovery (FLAIR) cervical spine MRI showing an area of increased signal B. Sagittal T1-weighted cervical spine MRI following gadolinium-diethylene triamine brain MRI following gadolinium DPTA infusion shows enhancement of the right optic within the area postrema (arrow). F. Axial T1-weighted brain MRI following — Sagittal T1-weighted cervical spine MRI following gadolinium-diethylene triamine pentaacetic acid (DPTA) infusion showing enhancement of the myelitis lesion.
Figure 3¶

Caption: FIGURE 456-1 Imaging findings in neuromyelitis optica: longitudinally extensive inversion recovery (FLAIR) cervical spine MRI showing an area of increased signal B. Sagittal T1-weighted cervical spine MRI following gadolinium-diethylene triamine brain MRI following gadolinium DPTA infusion shows enhancement of the right optic within the area postrema (arrow). F. Axial T1-weighted brain MRI following — Axial and coronal brain MRI following gadolinium DPTA infusion showing enhancement of the right optic nerve, indicative of optic neuritis.
Figure 4¶

Caption: FIGURE 456-1 Imaging findings in neuromyelitis optica: longitudinally extensive inversion recovery (FLAIR) cervical spine MRI showing an area of increased signal B. Sagittal T1-weighted cervical spine MRI following gadolinium-diethylene triamine brain MRI following gadolinium DPTA infusion shows enhancement of the right optic within the area postrema (arrow). F. Axial T1-weighted brain MRI following — Axial brain MRI showing an area of hyperintense signal on T2-weighted imaging within the area postrema (arrow), associated with area postrema syndrome.
Figure 5¶
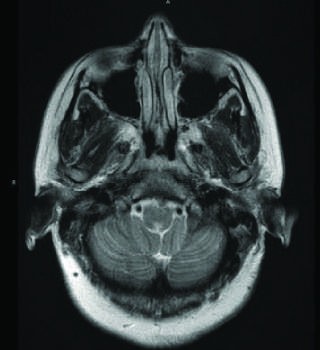
Caption: FIGURE 456-1 Imaging findings in neuromyelitis optica: longitudinally extensive inversion recovery (FLAIR) cervical spine MRI showing an area of increased signal B. Sagittal T1-weighted cervical spine MRI following gadolinium-diethylene triamine brain MRI following gadolinium DPTA infusion shows enhancement of the right optic within the area postrema (arrow). F. Axial T1-weighted brain MRI following — Axial T1-weighted brain MRI following gadolinium-DPTA infusion showing punctate enhancement of the area postrema (arrow).
Figure 6¶

Caption: FIGURE 456-1 Imaging findings in neuromyelitis optica: longitudinally extensive inversion recovery (FLAIR) cervical spine MRI showing an area of increased signal B. Sagittal T1-weighted cervical spine MRI following gadolinium-diethylene triamine brain MRI following gadolinium DPTA infusion shows enhancement of the right optic within the area postrema (arrow). F. Axial T1-weighted brain MRI following — Composite imaging findings in neuromyelitis optica demonstrating the spectrum of longitudinally extensive transverse myelitis, optic neuritis, and brainstem involvement.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.