Genetic Cardiomyopathies¶
Chapter 267 | Part 6: Disorders of the Cardiovascular System · Part 6 – Cardiovascular Disorders
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- Hypertrophic cardiomyopathy (HCM) prevalence is approximately 1:500 in North America, Africa, and Asia.
- Truncating variants in TTN (titin) are the most common genetic cause of dilated cardiomyopathy (DCM), accounting for up to 25% of familial disease.
- Sudden death risk factors for HCM include history of cardiac arrest, family history of sudden death, LV thickness >30 mm, and LV apical aneurysm.
- Fabry's disease is an X-linked disorder caused by deficiency of alpha-galactosidase A, leading to glycosphingolipid accumulation.
- In HCM, outflow tract obstruction is present in ~30% of patients at rest and can be provoked by exercise in another ~30%.
- Genetic testing utility is primarily to inform family evaluations, though it enables detection of specific therapies for metabolic disorders like Fabry's and Danon's disease.
- Athlete's heart hypertrophy regresses with cessation of training, unlike HCM, and is accompanied by supernormal exercise capacity (VO2max >50 mL/kg per min).
- LMNA variants in DCM warrant ICD placement before LVEF declines to 0.35 due to high arrhythmia risk.
- Danon disease presents with extreme LV hypertrophy in childhood, often progressing to end-stage heart failure with low ejection fraction.
- Barth's syndrome (TAZ variants) presents with skeletal myopathy, cognitive impairment, and neutropenia alongside DCM/LVNC.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- Initial Evaluation of Cardiomyopathy
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- Selected Genetic Defects Associated with Cardiomyopathy
- 4. CLINICAL FEATURES
- Hypertrophic Cardiomyopathy Clinical Presentation
- Dilated Cardiomyopathy Clinical Presentation
- Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
- Metabolic Cardiomyopathies
- 5. DIFFERENTIAL DIAGNOSIS
- 6. INVESTIGATIONS & DIAGNOSIS
- Risk Stratification for Sudden Death in Hypertrophic Cardiomyopathy
- 7. MANAGEMENT & TREATMENT
- Hypertrophic Cardiomyopathy Treatment Algorithm
- Dilated Cardiomyopathy Management
- Metabolic Cardiomyopathy Management
- 8. PROGNOSIS & COMPLICATIONS
- 9. SPECIAL CONSIDERATIONS
- 10. KEY PEARLS & CLINICAL TRAPS
- Flowcharts & Algorithms
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🔀 Flowchart | Treatment algorithm for hypertrophic cardiomyopathy depending on the with obstruction to outflow |
| 1 | 🖼 Figure | Drawing of myocyte indicating multiple sites of abnormal gene products proteins (actin,... |
| 2 | 🖼 Figure | Fabry’s disease |
| 3 | 🖼 Figure | Dilated cardiomyopathy |
| 4 | 🖼 Figure | Hypertrophic cardiomyopathy |
| 5 | 🖼 Figure | Dilated cardiomyopathy |
| 6 | 🖼 Figure | Hypertrophic cardiomyopathy |
| 7 | 🖼 Figure | Hypertrophic cardiomyopathy |
| 8 | 🖼 Figure | Dilated cardiomyopathy |
| 9 | 🖼 Figure | Figure / Illustration |
1. DEFINITION & OVERVIEW¶
- Genetic cardiomyopathy is characterized by age-dependent and incomplete penetrance.
- The defining phenotype of cardiomyopathy is rarely present at birth and, in some individuals, may never manifest.
- Related individuals who carry the same variant may differ in the severity and rate of progression of cardiac dysfunction and associated rhythm disorders.
- Harrison's defines genetic cardiomyopathy as a condition where each of the traditional morphologic forms of cardiomyopathy, hypertrophic, dilated, and restrictive, can be caused or modified by underlying genetic factors.
- Most familial cardiomyopathies are inherited in an autosomal dominant pattern, with occasional autosomal recessive, matrilineal (mitochondrial), and X-linked inheritance.
- Missense variants with amino acid substitutions and truncating variants are the most common genetic abnormalities in cardiomyopathy.
- Expressed mutant proteins may interfere with function of the normal allele through a dominant negative mechanism.
- Variants introducing a premature stop codon (nonsense) or shift in the reading frame (frameshift) may create a truncated or unstable protein, the lack of which causes cardiomyopathy (haploinsufficiency).
- In some forms of genetic DCM, both haploinsufficiency and the dominant negative effect of a truncated allele may contribute to pathophysiology.
- Deletions or duplications of an entire exon or gene are uncommon causes of cardiomyopathy, except for the dystrophinopathies.
- Many different genes have been implicated in human cardiomyopathy (locus heterogeneity), and many pathogenic or likely pathogenic variants within those genes have been associated with disease (allelic heterogeneity).
- While most identified variants are private to individual families, several specific variants are found repeatedly, either due to a founder effect or recurrent variants at a common residue.
- While most patients with genetic cardiomyopathy have a single rare, high effect, disease allele, there is a growing appreciation for the effect of multiple less rare intermediate effect alleles on penetrance and expression.
- The additive effects of multiple common alleles/variants captured as a polygenic risk score have shown greatest utility in common diseases such as atherosclerosis, but may also provide insight into the subset of patients with primary cardiomyopathy without an identifiable high effect allele.
- Currently, the greatest utility of genetic testing for cardiomyopathy is to inform family evaluations.
- Genetic testing can provide risk stratification in some forms of cardiomyopathy and occasionally enables the detection of a disease for which specific therapy is indicated, such as the replacements for defective metabolic enzymes in Fabry's disease and Gaucher's disease.
- Clinical trials are interrogating the role of gene therapies for cardiomyopathy, which will provide greater impetus for genetic testing.
Initial Evaluation of Cardiomyopathy¶
- Thorough history and physical examination to identify cardiac and noncardiac disorders.
- Detailed family history of heart failure, cardiomyopathy, skeletal myopathy, conduction disorders, tachyarrhythmias, and sudden death.
- History of alcohol, illicit drugs, chemotherapy or radiation therapy.
- Assessment of changing ability to perform routine and desired activities.
- Assessment of jugular venous pressure, edema, orthostatic blood pressure, adequacy of perfusion.
- Laboratory Evaluation: Electrocardiogram, Chest radiograph, Two-dimensional and Doppler echocardiogram, Magnetic resonance imaging for evidence of myocardial inflammation and fibrosis.
- Chemistry: Serum sodium, potassium, calcium, magnesium; Fasting glucose (glycohemoglobin in diabetes mellitus); Creatinine, blood urea nitrogen; Albumin, total protein, liver function tests; Lipid profile; Thyroid-stimulating hormone; Serum iron, transferrin saturation; Urinalysis; Creatine kinase isoforms; Cardiac troponin level.
- Hematology: Hemoglobin/hematocrit; White blood cell count with differential; Total eosinophil count if abnormal % on differential; Erythrocyte sedimentation rate.
- Evaluation When Specific Diagnoses Are Suspected: Respiratory pathogen panel during acute respiratory syndromes; Diagnosis of other specific infections such as Human immunodeficiency virus, Chagas' disease (Trypanosoma cruzi), Lyme disease (Borrelia burgdorferi) and other tick-borne diseases, Toxoplasmosis, Trichinosis.
- Genetic counseling and testing with multigene cardiomyopathy panel.
- Serologies for active rheumatologic disease.
- Endomyocardial biopsy including sample for electron microscopy when suspecting specific diagnosis with therapeutic implications.
- Catheterization with coronary angiography in patients who have evidence of ischemia/infarction and are candidates for intervention.
Table 1 — TABLE 266-2 Initial Evaluation of Cardiomyopathy¶
| Category | Evaluation Component | Details |
|---|---|---|
| Clinical Evaluation | Thorough history and physical examination | To identify cardiac and noncardiac disorders |
| Clinical Evaluation | Detailed family history | Heart failure, cardiomyopathy, skeletal myopathy, conduction disorders, tachyarrhythmias, and sudden death |
| Clinical Evaluation | History of exposures | Alcohol, illicit drugs, chemotherapy or radiation therapy |
| Clinical Evaluation | Functional assessment | Changing ability to perform routine and desired activities |
| Clinical Evaluation | Physical signs | Jugular venous pressure, edema, orthostatic blood pressure, adequacy of perfusion |
| Laboratory Evaluation | Electrocardiogram | Standard 12-lead ECG |
| Laboratory Evaluation | Chest radiograph | CXR |
| Laboratory Evaluation | Two-dimensional and Doppler echocardiogram | Structural and functional assessment |
| Laboratory Evaluation | Magnetic resonance imaging | Evidence of myocardial inflammation and fibrosis |
| Laboratory Evaluation | Chemistry | Serum sodium, potassium, calcium, magnesium; Fasting glucose; Creatinine, BUN; Albumin, total protein, liver function tests; Lipid profile; Thyroid-stimulating hormone; Serum iron, transferrin saturation; Urinalysis; Creatine kinase isoforms; Cardiac troponin level |
| Laboratory Evaluation | Hematology | Hemoglobin/hematocrit; White blood cell count with differential; Total eosinophil count if abnormal % on differential; Erythrocyte sedimentation rate |
| Evaluation When Specific Diagnoses Are Suspected | Infectious disease panel | Respiratory pathogen panel during acute respiratory syndromes; Diagnosis of other specific infections such as HIV, Chagas' disease, Lyme disease, Toxoplasmosis, Trichinosis |
| Evaluation When Specific Diagnoses Are Suspected | Genetic testing | Genetic counseling and testing with multigene cardiomyopathy panel |
| Evaluation When Specific Diagnoses Are Suspected | Autoimmune serology | Serologies for active rheumatologic disease |
| Evaluation When Specific Diagnoses Are Suspected | Biopsy | Endomyocardial biopsy including sample for electron microscopy when suspecting specific diagnosis with therapeutic implications |
| Evaluation When Specific Diagnoses Are Suspected | Angiography | Catheterization with coronary angiography in patients who have evidence of ischemia/infarction and are candidates for intervention |
2. EPIDEMIOLOGY¶
- Prevalence in North America, Africa, and Asia is about 1:500 for HCM.
- It is a leading cause of sudden death in the young and is an important cause of heart failure.
- Pediatric presentation is associated with increased early morbidity and mortality.
- Patients diagnosed as adults have decreased survival compared to age-matched individuals without hypertrophic cardiomyopathy.
- The recognized frequency of familial involvement in DCM has increased to >30%.
- Truncating variants in TTN, encoding the giant sarcomeric protein titin, are the most common cause of DCM, accounting for up to 25% of familial disease.
- On average, men with TTN truncating variants develop cardiomyopathy a decade before women, without distinctive clinical features.
- Variants in thick and thin filament genes account for ~8% of DCM and may manifest in early childhood.
- Familial clustering is present in ~30–40% of cases of DCM and arrhythmogenic cardiomyopathy (ACM).
- Monogenic etiologies can be identified in ~25% of DCM and ACM cases.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- Most identified variants are private to individual families, but several specific variants are found repeatedly, either due to a founder effect or recurrent variants at a common residue.
- While most patients with genetic cardiomyopathy have a single rare, high effect, disease allele, there is a growing appreciation for the effect of multiple less rare intermediate effect alleles on penetrance and expression.
- The additive effects of multiple common alleles/variants captured as a polygenic risk score have shown greatest utility in common diseases such as atherosclerosis, but may also provide insight into the subset of patients with primary cardiomyopathy without an identifiable high effect allele.
- Genetic cardiomyopathy is characterized by age-dependent and incomplete penetrance.
- The defining phenotype of cardiomyopathy is rarely present at birth and, in some individuals, may never manifest.
- Related individuals who carry the same variant may differ in the severity and rate of progression of cardiac dysfunction and associated rhythm disorders, indicating the important role of other genetic, epigenetic, and environmental modifiers in disease expression.
- Sex appears to play a role, as penetrance and clinical severity may be greater in men for most cardiomyopathies.
- The clinical course of a patient usually cannot be predicted based on which variant is present; thus, current therapy is based on the phenotype rather than the genetic defect.
Selected Genetic Defects Associated with Cardiomyopathy¶
- Sarcomere gene variants are present in ~40–50% of patients with hypertrophic cardiomyopathy and are more common in those with familial disease and characteristic asymmetric septal hypertrophy.
- More than nine different genes with >1500 variants have been implicated in HCM, although ~80% of patients have a variant in either MYH7 or MYBPC3.
- Truncating variants in TTN are the most common cause of DCM.
- Defects in the sarcolemmal membrane proteins are associated with DCM.
- The best known is dystrophin, encoded by the X chromosome gene DMD, abnormalities of which cause Duchenne's and Becker's muscle dystrophy.
- Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias.
- Variants in SCN5A, the alpha subunit of the Nav 1.5 ion channel protein, distinct from those that cause the Brugada or long QT syndromes, have been implicated in DCM with conduction disease.
- Nuclear membrane protein defects in cardiac and skeletal muscle occur in either autosomal (lamin A/C) or X-linked (emerin) patterns.
- Intercalated disks contribute to intracellular connections, allowing mechanical and electrical coupling between cells and also connections to desmin filaments within the cell.
- Variants in proteins of the desmosomal complex compromise attachment of the myocytes, which can become disconnected and die via activation of Wnt/beta-catenin and proinflammatory signaling pathways, to be replaced by fat and fibrous tissue.
- These areas are highly arrhythmogenic and may dilate to form aneurysms.
- Although more often noted in the right ventricle (arrhythmogenic right ventricular cardiomyopathy), this condition can be restricted to the left ventricle (especially when secondary to truncating variants in DSP, which encodes desmoplakin) or affect both ventricles and has also been termed arrhythmogenic cardiomyopathy.
- Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells.
- Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing pseudohypertrophy, usually with an abnormally short PR interval.
- The pseudohypertrophic phenotype is most common, but restrictive cardiomyopathy and DCM may occur.
- Most of these diseases are diagnosed during childhood.
Table 2 — TABLE 267-1 Selected Genetic Defects Associated with Cardiomyopathy¶
| Gene Product | Inheritance | Cardiac Phenotype | Isolated Cardiac Phenotype | Extracardiac Manifestations |
|---|---|---|---|---|
| ACTC1 (cardiac actin) | AD | HCM, DCM | Yes | |
| MYH7 (beta myosin heavy chain) | AD | HCM, DCM, LVNC | Yes | Skeletal myopathy |
| MYBPC3 (myosin binding protein C) | AD | HCM | Yes | |
| TNNT2 (cardiac troponin T) | AD | HCM, DCM, LVNC | Yes | |
| TNNI3 (cardiac troponin I) | AD, AR | HCM, DCM, RCM | Yes | |
| TTN (Titin) | AD | DCM | Yes | |
| TPM1 (alpha-tropomyosin) | AD | HCM, DCM | Yes | |
| TNNC1 (cardiac troponin C) | AD | DCM | Yes | |
| MYL2 (myosin regulatory light chain) | AD | HCM | Yes | Skeletal myopathy |
| MYL3 (myosin essential light chain) | AD | HCM | Yes | |
| DES (desmin) | AD | RCM, DCM | Yes | Skeletal myopathy |
| FLNC (filamin C) | AD | DCM | Yes | Skeletal myopathy |
| NEXN (nexilin) | AD | DCM | Yes | |
| VCL (vinculin) | AD | DCM | Yes | |
| LMNA (lamin A/C) | AD, AR | CDDC | Yes | Skeletal myopathy |
| EMD (emerin) | X-linked | CDDC | No | Skeletal myopathy, contractures |
| PLN (phospholamban) | AD | DCM, ARVC | Yes | |
| SCN5A (NAV 1.5) | AD | CDDC | Yes | Note other variants associated with Brugada syndrome |
| RYR2 (cardiac ryanodine receptor) | AD | ARVC | Yes | |
| CASQ2 (calsequestrin 2) | AR | ARVC | Yes | |
| PRKAG2 (gamma-subunit of AMP kinase) | AD | HCM+ | Yes | |
| LAMP2 (lysosomal associated membrane protein) | X-linked | HCM+ | No | Danon's disease: skeletal myopathy, cognitive impairment |
| TAZ (tafazzin) | X-linked | DCM, LVNC | No | Barth's syndrome: skeletal myopathy, cognitive impairment, neutropenia |
| FXN (frataxin) | AR | HCM | No | Friedreich's ataxia: ataxia, diabetes mellitus type 2 |
| TMEM43 (transmembrane protein 43) | AD | ARVC | Yes | |
| GLA (alpha-galactosidase-A) | X-linked | HCM+ | No | Fabry's disease: renal failure, angiokeratomas and painful neuropathy |
| Mitochondrial DNA | Maternal transmission | DCM, HCM | No | MELAS, MERRF, Kearns-Sayre syndrome, ocular myopathy |
| DMD (dystrophin) | X-linked | DCM | No | Duchenne's and Becker's muscular dystrophy |
| DMPK (dystrophica myotonica protein kinase) | AD | DCM | No | Myotonic dystrophy type 1 |
| DSP (desmoplakin), JUP (plakoglobin) | AD, AR | ARVC, DCM | Yes | Carvajal syndrome (AR), Naxos syndrome (AR), woolly hair and hyperkeratosis of palms and soles |
| DSG2 (desmoglein 2), DSC2 (desmocollin 2), PKP2 (plakophilin 2) | AD | ARVC | Yes | |
| RBM20 (RNA binding motif 20) | AD | DCM | Yes | |
| BAG3 (BCL2-associated athanogene 3) | AD | DCM | Yes | |
| ALPK3 (alpha-kinase 3) | AR | HCM | Yes |
4. CLINICAL FEATURES¶
- Hypertrophic cardiomyopathy is defined as left ventricular hypertrophy that develops in the absence of causative hemodynamic factors, such as hypertension, aortic valve disease, or systemic infiltrative or storage diseases.
- It has previously been termed hypertrophic obstructive cardiomyopathy (HOCM); however, the accepted terminology is now hypertrophic cardiomyopathy with or without obstruction.
- Pediatric presentation is associated with increased early morbidity and mortality.
- Patients diagnosed as adults have decreased survival compared to age-matched individuals without hypertrophic cardiomyopathy.
- Women appear to have lower penetrance of sarcomere variants and an older age at hypertrophic cardiomyopathy diagnosis but subsequently increased rates of heart failure and mortality thereafter.
- In MYBPC3 variant carriers, the average age of disease development is ~40 years, while 30% remain free from hypertrophy after 70 years.
- Related individuals who carry the same variant may have a different extent and pattern of hypertrophy (e.g., asymmetric vs concentric), occurrence of outflow tract obstruction, and associated clinical outcomes.
- Sudden death and progression to heart failure occur more commonly in families with that history.
- At the level of the sarcomere, hypertrophic cardiomyopathy variants lead to enhanced calcium sensitivity, maximal force generation, and ATPase activity.
- Calcium handling is affected through modification of regulatory proteins.
- Sarcomere variants lead to abnormal energetics and impaired relaxation, both directly and as a result of hypertrophy.
- Hypertrophic cardiomyopathy is characterized by misalignment and disarray of the enlarged myofibrils and myocytes, which can also occur to a lesser extent in other cardiac diseases.
- Although hypertrophy is the defining feature of hypertrophic cardiomyopathy, fibrosis and microvascular disease are also present.
- Interstitial fibrosis is detectable before overt hypertrophy develops and likely results from early activation of profibrotic pathways.
- In the majority of patients with overt cardiomyopathy, focal areas of replacement fibrosis can be readily detected with magnetic resonance imaging (MRI).
- These areas of scar may represent substrate for the development of ventricular arrhythmias.
- Increased thickness and decreased luminal area of the intramural vessels in hypertrophied myocardium contribute to microvascular ischemia and angina.
- Microinfarction of hypertrophied myocardium is a hypothesized mechanism for replacement scar formation.
- Macroscopically, hypertrophy is typically manifest as nonuniform hypertrophy.
- The interventricular septum is the typical location of maximal hypertrophy, although other patterns of hypertrophic remodeling include concentric and midventricular.
- Hypertrophy confined to the ventricular apex (apical hypertrophic cardiomyopathy) is less often familial and has a different genetic substrate, with sarcomere variants present in only ~15%.
- Left ventricular outflow tract obstruction represents the most common focus of diagnosis and intervention.
- Diastolic dysfunction, myocardial fibrosis, and microvascular ischemia also contribute to contractile dysfunction and elevated intracardiac pressures.
- Obstruction is present in ~30% of patients at rest and can be provoked by exercise in another ~30%.
- Systolic obstruction is initiated by drag forces, which push an anteriorly displaced and enlarged anterior mitral leaflet into contact with the hypertrophied ventricular septum.
- Mitral leaflet coaptation may ensue, leading to posteriorly directed mitral regurgitation.
- To maintain stroke volume across outflow tract obstruction, the ventricle generates higher pressures, leading to higher wall stress and myocardial oxygen demand.
- Smaller chamber size and increased contractility exacerbate the severity of obstruction.
- Conditions of low preload, such as dehydration, and low afterload, such as arterial vasodilation, may lead to transient hypotension and near-syncope.
- The systolic ejection murmur of left ventricular outflow tract obstruction is harsh and late peaking and can be enhanced by bedside maneuvers that diminish ventricular volume and transiently worsen obstruction, such as the Valsalva maneuver or standing from a squatting position.
- Patients may be diagnosed after undergoing evaluations triggered by the abnormal physical findings (murmur) or by symptoms of exertional dyspnea, angina, or syncope.
- Alternatively, diagnosis may follow evaluations prompted by the detection of disease in family members.
- Rigorous athletic training (athlete's heart) may cause intermediate degrees of physiologic hypertrophy difficult to differentiate from mild hypertrophic cardiomyopathy.
- Unlike hypertrophic cardiomyopathy, hypertrophy in the athlete's heart regresses with cessation of training and is accompanied by supernormal exercise capacity (VO2max >50 mL/kg per min), mild ventricular dilation, and normal diastolic function.
- An enlarged left ventricle with reduced systolic function as measured by LVEF characterizes DCM.
- DCM is considered to be present when the LVEF is ≤0.50 and/or the left ventricular diastolic dimension is >95% predicted for age and sex.
- In some patients with reduced LVEF, the left ventricular dilation is minimal, sometimes referred to as nondilated or minimally dilated cardiomyopathy.
- While diverse etiologies may cause DCM and arrhythmogenic cardiomyopathy (ACM), familial clustering is present in ~30–40% of cases.
- Monogenic etiologies can be identified in ~25% of DCM and ACM cases.
- Sarcomere variants are most associated with hypertrophic cardiomyopathy; however, they are also implicated in DCM.
- The most common genetic causes of DCM are truncating variants of the giant protein titin, encoded by TTN, which maintains sarcomere structure and acts as a key signaling molecule.
- Defects in the sarcolemmal membrane proteins are associated with DCM.
- The best known is dystrophin, encoded by the X chromosome gene DMD, abnormalities of which cause Duchenne's and Becker's muscle dystrophy.
- This protein provides a network that supports the sarcolemma and also connects to the sarcomere.
- The progressive functional defect in both cardiac and skeletal muscle reflects vulnerability to mechanical stress.
- Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias.
- Variants in SCN5A, the alpha subunit of the Nav 1.5 ion channel protein, distinct from those that cause the Brugada or long QT syndromes, have been implicated in DCM with conduction disease.
- Nuclear membrane protein defects in cardiac and skeletal muscle occur in either autosomal (lamin A/C) or X-linked (emerin) patterns.
- These defects are associated with a high prevalence of atrial and ventricular arrhythmias and conduction system disease, which can occur in some family members without or before detectable cardiomyopathy and underlie gene-specific risk stratification for sudden death and different utilization criteria for primary prevention implantable cardioverter-defibrillators in these patients.
- Intercalated disks contribute to intracellular connections, allowing mechanical and electrical coupling between cells and also connections to desmin filaments within the cell.
- Variants in proteins of the desmosomal complex compromise attachment of the myocytes, which can become disconnected and die via activation of Wnt/beta-catenin and proinflammatory signaling pathways, to be replaced by fat and fibrous tissue.
- These areas are highly arrhythmogenic and may dilate to form aneurysms.
- Although more often noted in the right ventricle (arrhythmogenic right ventricular cardiomyopathy), this condition can be restricted to the left ventricle (especially when secondary to truncating variants in DSP, which encodes desmoplakin) or affect both ventricles and has also been termed arrhythmogenic cardiomyopathy.
- Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells.
- Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing pseudohypertrophy, usually with an abnormally short PR interval.
- The pseudohypertrophic phenotype is most common, but restrictive cardiomyopathy and DCM may occur.
- Most of these diseases are diagnosed during childhood.
- Fabry's disease results from a deficiency of the lysosomal enzyme alpha-galactosidase A caused by variants in GLA.
- This disorder of glycosphingolipid metabolism is an X-linked disorder that may also cause clinical disease in female carriers.
- Glycolipid accumulation may be limited to the cardiac tissues but usually also involves the skin, peripheral nerve, and kidney.
- Electron microscopy of endomyocardial biopsy tissue shows diagnostic vesicles containing concentric lamellar figures.
- Diagnosis can be made through assessment of enzyme activity and/or GLA.
- Most restrictive cardiomyopathy (RCM) is due to acquired causes, and there is increasing emphasis to diagnose amyloidosis due to transthyretin variants.
- Inherited metabolic and storage diseases can cause RCM, as can variants in DES causing combined cardiac and skeletal myopathy and sarcomere variants causing an overlap of RCM and hypertrophic cardiomyopathy.
Hypertrophic Cardiomyopathy Clinical Presentation¶
- Patients may be diagnosed after undergoing evaluations triggered by the abnormal physical findings (murmur) or by symptoms of exertional dyspnea, angina, or syncope.
- Alternatively, diagnosis may follow evaluations prompted by the detection of disease in family members.
- Cardiac imaging is central to diagnosis, for which the physical examination and ECG are insensitive.
- The identification of a disease-causing variant in a proband can focus family evaluations on variant carriers, but this strategy requires a high degree of certainty that the variant is truly pathogenic and not a benign DNA variant.
- Biopsy is not recommended to diagnose hypertrophic cardiomyopathy but can be used to exclude infiltrative and metabolic diseases.
Dilated Cardiomyopathy Clinical Presentation¶
- An enlarged left ventricle with reduced systolic function as measured by LVEF characterizes DCM.
- DCM is considered to be present when the LVEF is ≤0.50 and/or the left ventricular diastolic dimension is >95% predicted for age and sex.
- In some patients with reduced LVEF, the left ventricular dilation is minimal, sometimes referred to as nondilated or minimally dilated cardiomyopathy.
- While diverse etiologies may cause DCM and arrhythmogenic cardiomyopathy (ACM), familial clustering is present in ~30–40% of cases.
- Monogenic etiologies can be identified in ~25% of DCM and ACM cases.
- Sarcomere variants are most associated with hypertrophic cardiomyopathy; however, they are also implicated in DCM.
- The most common genetic causes of DCM are truncating variants of the giant protein titin, encoded by TTN, which maintains sarcomere structure and acts as a key signaling molecule.
- Defects in the sarcolemmal membrane proteins are associated with DCM.
- The best known is dystrophin, encoded by the X chromosome gene DMD, abnormalities of which cause Duchenne's and Becker's muscle dystrophy.
- This protein provides a network that supports the sarcolemma and also connects to the sarcomere.
- The progressive functional defect in both cardiac and skeletal muscle reflects vulnerability to mechanical stress.
- Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias.
- Variants in SCN5A, the alpha subunit of the Nav 1.5 ion channel protein, distinct from those that cause the Brugada or long QT syndromes, have been implicated in DCM with conduction disease.
- Nuclear membrane protein defects in cardiac and skeletal muscle occur in either autosomal (lamin A/C) or X-linked (emerin) patterns.
- These defects are associated with a high prevalence of atrial and ventricular arrhythmias and conduction system disease, which can occur in some family members without or before detectable cardiomyopathy and underlie gene-specific risk stratification for sudden death and different utilization criteria for primary prevention implantable cardioverter-defibrillators in these patients.
- Intercalated disks contribute to intracellular connections, allowing mechanical and electrical coupling between cells and also connections to desmin filaments within the cell.
- Variants in proteins of the desmosomal complex compromise attachment of the myocytes, which can become disconnected and die via activation of Wnt/beta-catenin and proinflammatory signaling pathways, to be replaced by fat and fibrous tissue.
- These areas are highly arrhythmogenic and may dilate to form aneurysms.
- Although more often noted in the right ventricle (arrhythmogenic right ventricular cardiomyopathy), this condition can be restricted to the left ventricle (especially when secondary to truncating variants in DSP, which encodes desmoplakin) or affect both ventricles and has also been termed arrhythmogenic cardiomyopathy.
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)¶
- Arrhythmogenic right ventricular cardiomyopathy (ARVC) has an established diagnostic framework (task force criteria) that rests upon identifying and quantifying right ventricular dilation, dyskinesia, and ECG abnormalities (repolarization, depolarization, and arrhythmias) in the context of a suggestive family history of genetic test results.
- Overlapping with ARVC and DCM is ACM, in which ventricular arrhythmias may precede or supersede the severity of predominantly left ventricular remodeling.
- Unlike ARVC, consensus diagnostic criteria for ACM are lacking.
- Early stages of ARVC and ACM may be restricted to ventricular arrhythmias, and over years, ventricular dilation, hypokinesis, and failure may ensue.
- Patients with an initial presentation of right ventricular cardiomyopathy who progress to include left ventricular dysfunction are at high risk for adverse events.
Metabolic Cardiomyopathies¶
- Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells.
- Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing pseudohypertrophy, usually with an abnormally short PR interval.
- The pseudohypertrophic phenotype is most common, but restrictive cardiomyopathy and DCM may occur.
- Most of these diseases are diagnosed during childhood.
- Fabry's disease results from a deficiency of the lysosomal enzyme alpha-galactosidase A caused by variants in GLA.
- This disorder of glycosphingolipid metabolism is an X-linked disorder that may also cause clinical disease in female carriers.
- Glycolipid accumulation may be limited to the cardiac tissues but usually also involves the skin, peripheral nerve, and kidney.
- Electron microscopy of endomyocardial biopsy tissue shows diagnostic vesicles containing concentric lamellar figures.
- Diagnosis can be made through assessment of enzyme activity and/or GLA.
- Danon disease presents with extreme left ventricular hypertrophy appearing early, often in childhood, and can progress rapidly to end-stage heart failure with low ejection fraction.
- Electron microscopy of these metabolic disorders shows that the myocytes are enlarged by multiple intracellular vacuoles of metabolic by-products.
- Gene therapy using a viral vector to deliver functional LAMP2 to cardiomyocytes is currently under study in humans with Danon disease.
- Barth's syndrome presents with skeletal myopathy, cognitive impairment, and neutropenia alongside DCM/LVNC.
5. DIFFERENTIAL DIAGNOSIS¶
- Rigorous athletic training (athlete's heart) may cause intermediate degrees of physiologic hypertrophy difficult to differentiate from mild hypertrophic cardiomyopathy.
- Unlike hypertrophic cardiomyopathy, hypertrophy in the athlete's heart regresses with cessation of training and is accompanied by supernormal exercise capacity (VO2max >50 mL/kg per min), mild ventricular dilation, and normal diastolic function.
- Most restrictive cardiomyopathy (RCM) is due to acquired causes, and there is increasing emphasis to diagnose amyloidosis due to transthyretin variants.
- Inherited metabolic and storage diseases can cause RCM, as can variants in DES causing combined cardiac and skeletal myopathy and sarcomere variants causing an overlap of RCM and hypertrophic cardiomyopathy.
- Biopsy is not recommended to diagnose hypertrophic cardiomyopathy but can be used to exclude infiltrative and metabolic diseases.
6. INVESTIGATIONS & DIAGNOSIS¶
- For any patient with suspected or proven genetic disease, family members should be considered and evaluated in a longitudinal fashion.
- Screening generally includes both an echocardiogram and electrocardiogram (ECG).
- The indications and implications for confirmatory specific genetic testing vary depending on the specific variant.
- The identification of a disease-causing variant in a proband can focus family evaluations on variant carriers, but this strategy requires a high degree of certainty that the variant is truly pathogenic and not a benign DNA variant.
- Biopsy is not recommended to diagnose hypertrophic cardiomyopathy but can be used to exclude infiltrative and metabolic diseases.
- Rigorous athletic training (athlete's heart) may cause intermediate degrees of physiologic hypertrophy difficult to differentiate from mild hypertrophic cardiomyopathy.
- Unlike hypertrophic cardiomyopathy, hypertrophy in the athlete's heart regresses with cessation of training and is accompanied by supernormal exercise capacity (VO2max >50 mL/kg per min), mild ventricular dilation, and normal diastolic function.
- The defining phenotype of cardiomyopathy is rarely present at birth and, in some individuals, may never manifest.
- Related individuals who carry the same variant may differ in the severity and rate of progression of cardiac dysfunction and associated rhythm disorders, indicating the important role of other genetic, epigenetic, and environmental modifiers in disease expression.
- Sex appears to play a role, as penetrance and clinical severity may be greater in men for most cardiomyopathies.
- The clinical course of a patient usually cannot be predicted based on which variant is present; thus, current therapy is based on the phenotype rather than the genetic defect.
- Currently, the greatest utility of genetic testing for cardiomyopathy is to inform family evaluations.
- However, genetic testing can provide risk stratification in some forms of cardiomyopathy and occasionally enables the detection of a disease for which specific therapy is indicated, such as the replacements for defective metabolic enzymes in Fabry's disease and Gaucher's disease.
- Moreover, clinical trials are interrogating the role of gene therapies for cardiomyopathy, which will provide greater impetus for genetic testing.
Risk Stratification for Sudden Death in Hypertrophic Cardiomyopathy¶
- Patients with hypertrophic cardiomyopathy have an increased risk of sudden cardiac death from ventricular tachyarrhythmias.
- Vigorous physical activity and competitive sports have been historically prohibited; however, recent studies have failed to identify associated relationship between exertion and ventricular arrhythmias in hypertrophic cardiomyopathy, empowering patients and providers to make shared decisions about exercise.
- Factors that increase the risk of sudden death from a baseline of 0.5% per year are presented in Table 267-2.
- As sudden death has not been reduced by medical or procedural interventions, traditionally an implantable cardioverter-defibrillator has been advised for patients with one or more major risk factors and advised on a selected basis for patient with more than one modifying risk factor.
- Nevertheless, the positive predictive value of most risk factors is low, and many patients receiving a defibrillator never receive an appropriate device therapy.
- A complementary approach to sudden death risk stratification and discussion with patients is the application of an externally validated European Society of Cardiology risk score using major criteria from Table 267-2 and continuous variables such as outflow tract gradient and left atrial size.
- Shared decision-making around implantable cardioverter-defibrillator implantation for primary prevention has emphasized discussions of estimated risk levels rather than dichotomous yes–no criteria.
- Long-term use of a defibrillator may be associated with serious device-related complications, particularly in young active patients.
- Refinement of sudden death risk through the application of contemporary technologies such as cardiac MRI is ongoing.
- Atrial fibrillation is common in patients with hypertrophic cardiomyopathy and may lead to hemodynamic deterioration and embolic stroke.
- Rapid ventricular response is poorly tolerated and may worsen outflow tract obstruction.
- Beta-adrenergic blocking agents and L-type calcium channel blockers slow atrioventricular (AV) nodal conduction and improve symptoms; cardiac glycosides should be avoided, as they may increase contractility and worsen obstruction.
- Even with adequate rate control, symptoms exacerbated by atrial fibrillation may persist due to loss of AV synchrony and may require restoration of sinus rhythm.
- Disopyramide and amiodarone are the preferred antiarrhythmic agents, with radiofrequency ablation considered for medically refractory cases.
- Anticoagulation to prevent embolic stroke in atrial fibrillation is recommended.
Table 3 — TABLE 267-2 Risk Stratification for Sudden Death in Hypertrophic Cardiomyopathy¶
| Major Risk Factor | Screening Technique | Details |
|---|---|---|
| History of cardiac arrest or spontaneous sustained ventricular tachycardia | History | Nonvagal, often with or after exertion |
| Family history of sudden cardiac death | Family history | Generally applicable to patients with apical hypertrophy |
| LV thickness >30 mm | Echocardiography or cardiac magnetic resonance imaging | Present in <10% of patients |
| LV apical aneurysm | Echocardiography or cardiac magnetic resonance imaging | Present in <10% of patients |
| LV systolic dysfunction (ejection fraction <50%) | Echocardiography or cardiac magnetic resonance imaging | Present in <10% of patients |
| LV outflow tract gradient | Echocardiography | Peak gradient measured at rest or with the Valsalva maneuver, mmHg |
| Left atrial diameter | Echocardiography | Diameter measured in the parasternal long axis, mm |
| LV thickness | Echocardiography | Maximal wall thickness, mm |
| Syncope, family history, nonsustained ventricular tachycardia | As above | As above |
| Spontaneous nonsustained ventricular tachycardia | Exercise or 24-h to 48-h ambulatory recording | >3 beats at rate >120 |
Table 4 — Variables Utilized in the European Society of Calculator for Estimated Risk of Sudden Death¶
| Variable | Measurement | Screening Technique |
|---|---|---|
| LV outflow tract gradient | Peak gradient measured at rest or with the Valsalva maneuver, mmHg | Echocardiography |
| Left atrial diameter | Diameter measured in the parasternal long axis, mm | Echocardiography |
| LV thickness | Maximal wall thickness, mm | Echocardiography |
| Syncope, family history, nonsustained ventricular tachycardia | As above | As above |
| Spontaneous nonsustained ventricular tachycardia | >3 beats at rate >120 | Exercise or 24-h to 48-h ambulatory recording |
7. MANAGEMENT & TREATMENT¶
- Management focuses on treatment of symptoms and prevention of sudden death and stroke.
- Left ventricular outflow tract obstruction can be controlled medically in the majority of patients.
- Beta-adrenergic blocking agents and L-type calcium channel blockers (e.g., verapamil) are first-line agents that reduce the severity of obstruction by slowing heart rate, enhancing diastolic filling, and decreasing contractility.
- Persistent symptoms of exertional dyspnea or chest pain can be controlled occasionally with the addition of disopyramide, an antiarrhythmic agent with potent negative inotropic properties.
- The recently introduced small-molecule cardiac myosin inhibitors mavacamten (U.S. Food and Drug Administration approved) and aficamten (under investigation) have shown high efficacy in the treatment of symptomatic obstructive hypertrophic cardiomyopathy, including in patients with persistent symptoms despite treatment with a beta blocker and/or those with persistent symptoms despite treatment with a beta blocker and/or calcium channel blocker.
- Patients with or without obstruction may develop heart failure symptoms due to fluid retention and require diuretic therapies for venous congestion.
- Severe medically refractory symptoms develop in ~5% of patients, for whom septal reduction therapy with surgical myectomy or alcohol septal ablation may be effective.
- Developed over 60 years ago, surgical myectomy effectively relieves outflow tract obstruction by excising part of the septal myocardium involved in the dynamic obstruction.
- In selected patients, perioperative mortality is extremely low with excellent long-term survival free from recurrent obstruction and symptoms.
- Mitral valve repair or replacement is usually unnecessary as associated eccentric mitral regurgitation resolves with myectomy alone.
- Alcohol septal ablation in patients with suitable coronary anatomy can relieve outflow tract obstruction via a controlled infarction of the proximal septum, which produces similar perioperative outcomes and gradient reduction as surgical myectomy.
- Although head-to-head comparisons between the two septal reduction therapies do not exist, septal ablation is relegated primarily to patients who wish to avoid surgery or who have limiting comorbidities.
- Neither procedure has been shown to improve outcomes other than symptoms.
- With both procedures, the most common complication is the development of complete heart block necessitating permanent pacing.
- However, ventricular pacing as a primary therapy for outflow tract obstruction is ineffective and not generally advised.
- As for HCM, shared decision-making is needed regarding implantable cardioverter-defibrillators.
- However, the specific genotype plays a greater role in decisions regarding DCM, for which some features may warrant implantable cardioverter-defibrillator placement before the LVEF has declined to 0.35, such as the presence of pathogenic LMNA or FLNC variants or a family history of sudden death at early ages.
- Prognosis and therapy of DCM and ACM are dictated primarily by the stage of clinical disease and the risk for sudden death, which also varies based on disease gene (higher for patients with variants in DES, DSP, DSC2, DCG2, FLNC, LMNA, PKP2, PLN, RBM20, SCN5A, and TMEM43).
- The rate of progression of disease is also heritable, with marked variation based on disease gene observed (e.g., patients with TTN truncating variants frequently experience recovery with medical therapy).
- Medical therapy is generally guided by the phenotype, such that patients with DCM and reduced LVEF are treated with therapies recommended for heart failure with reduced ejection fraction.
- As for HCM, shared decision-making is needed regarding implantable cardioverter-defibrillators.
- However, the specific genotype plays a greater role in decisions regarding DCM, for which some features may warrant implantable cardioverter-defibrillator placement before the LVEF has declined to 0.35, such as the presence of pathogenic LMNA or FLNC variants or a family history of sudden death at early ages.
- Arrhythmogenic right ventricular cardiomyopathy (ARVC) has an established diagnostic framework (task force criteria) that rests upon identifying and quantifying right ventricular dilation, dyskinesia, and ECG abnormalities (repolarization, depolarization, and arrhythmias) in the context of a suggestive family history of genetic test results.
- Overlapping with ARVC and DCM is ACM, in which ventricular arrhythmias may precede or supersede the severity of predominantly left ventricular remodeling.
- Unlike ARVC, consensus diagnostic criteria for ACM are lacking.
- Early stages of ARVC and ACM may be restricted to ventricular arrhythmias, and over years, ventricular dilation, hypokinesis, and failure may ensue.
- Patients with an initial presentation of right ventricular cardiomyopathy who progress to include left ventricular dysfunction are at high risk for adverse events.
- Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells.
- Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing pseudohypertrophy, usually with an abnormally short PR interval.
- The pseudohypertrophic phenotype is most common, but restrictive cardiomyopathy and DCM may occur.
- Most of these diseases are diagnosed during childhood.
- Fabry's disease results from a deficiency of the lysosomal enzyme alpha-galactosidase A caused by variants in GLA.
- This disorder of glycosphingolipid metabolism is an X-linked disorder that may also cause clinical disease in female carriers.
- Glycolipid accumulation may be limited to the cardiac tissues but usually also involves the skin, peripheral nerve, and kidney.
- Electron microscopy of endomyocardial biopsy tissue shows diagnostic vesicles containing concentric lamellar figures.
- Diagnosis can be made through assessment of enzyme activity and/or GLA.
- Most restrictive cardiomyopathy (RCM) is due to acquired causes, and there is increasing emphasis to diagnose amyloidosis due to transthyretin variants.
- Inherited metabolic and storage diseases can cause RCM, as can variants in DES causing combined cardiac and skeletal myopathy and sarcomere variants causing an overlap of RCM and hypertrophic cardiomyopathy.
- Gene therapy using a viral vector to deliver functional LAMP2 to cardiomyocytes is currently under study in humans with Danon disease.
Hypertrophic Cardiomyopathy Treatment Algorithm¶
- In all patients, evaluate risk for sudden death.
- If high, consider ICD.
- If low, follow with serial evaluation.
- Titrate beta blocker and/or calcium channel blocker.
- Use diuretics with caution to avoid hypovolemia, particularly in presence of symptoms.
- If evidence of outflow gradient:
- No: Try mavacamten or disopyramide.
- Yes: Consider procedure (Septal ablation or Septal myectomy).
- If evidence of severe progressive LV dysfunction or disarray:
- No: Reevaluate cause of symptoms.
- Yes: Consider cardiac transplantation.
- If refractory severe symptoms:
- Consider procedure (Septal ablation or Septal myectomy).
- Rarely, consider cardiac transplantation.
Table 5 — FIGURE 267-5 Treatment Algorithm for Hypertrophic Cardiomyopathy¶
| Step | Decision | Action |
|---|---|---|
| 1 | Evaluate risk for sudden death | All patients with HCM should be evaluated for atrial fibrillation and risk of sudden death, whether or not they require treatment for symptoms. |
| 2 | Symptomatic? | No: Evaluate risk for sudden death. Yes: Titrate beta blocker and/or calcium channel blocker. |
| 3 | Evidence of outflow gradient? | No: Try mavacamten or disopyramide. Yes: Consider procedure (Septal ablation or Septal myectomy). |
| 4 | Evidence of severe progressive LV dysfunction or disarray? | No: Reevaluate cause of symptoms. Yes: Consider cardiac transplantation. |
| 5 | Refractory severe symptoms? | Consider procedure (Septal ablation or Septal myectomy). Rarely, consider cardiac transplantation. |
Dilated Cardiomyopathy Management¶
- Prognosis and therapy of DCM and ACM are dictated primarily by the stage of clinical disease and the risk for sudden death, which also varies based on disease gene (higher for patients with variants in DES, DSP, DSC2, DCG2, FLNC, LMNA, PKP2, PLN, RBM20, SCN5A, and TMEM43).
- The rate of progression of disease is also heritable, with marked variation based on disease gene observed (e.g., patients with TTN truncating variants frequently experience recovery with medical therapy).
- Medical therapy is generally guided by the phenotype, such that patients with DCM and reduced LVEF are treated with therapies recommended for heart failure with reduced ejection fraction.
- As for HCM, shared decision-making is needed regarding implantable cardioverter-defibrillators.
- However, the specific genotype plays a greater role in decisions regarding DCM, for which some features may warrant implantable cardioverter-defibrillator placement before the LVEF has declined to 0.35, such as the presence of pathogenic LMNA or FLNC variants or a family history of sudden death at early ages.
Metabolic Cardiomyopathy Management¶
- Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells.
- Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing pseudohypertrophy, usually with an abnormally short PR interval.
- The pseudohypertrophic phenotype is most common, but restrictive cardiomyopathy and DCM may occur.
- Most of these diseases are diagnosed during childhood.
- Fabry's disease results from a deficiency of the lysosomal enzyme alpha-galactosidase A caused by variants in GLA.
- This disorder of glycosphingolipid metabolism is an X-linked disorder that may also cause clinical disease in female carriers.
- Glycolipid accumulation may be limited to the cardiac tissues but usually also involves the skin, peripheral nerve, and kidney.
- Electron microscopy of endomyocardial biopsy tissue shows diagnostic vesicles containing concentric lamellar figures.
- Diagnosis can be made through assessment of enzyme activity and/or GLA.
- Gene therapy using a viral vector to deliver functional LAMP2 to cardiomyocytes is currently under study in humans with Danon disease.
8. PROGNOSIS & COMPLICATIONS¶
- The general prognosis for hypertrophic cardiomyopathy is better than in early studies of referral populations, but mortality remains higher than in an age-matched population without cardiomyopathy.
- The sudden death risk is <1% per year; however, up to 1 in 20 patients will progress to overt systolic dysfunction with a reduced ejection fraction (<50%) with or without dilated remodeling (i.e., burned out or end-stage stage hypertrophic cardiomyopathy).
- These patients may suffer from progressive heart failure and sudden death unless they undergo timely cardiac transplantation.
- While diverse etiologies may cause DCM and arrhythmogenic cardiomyopathy (ACM), familial clustering is present in ~30–40% of cases.
- Monogenic etiologies can be identified in ~25% of DCM and ACM cases.
- Sarcomere variants are most associated with hypertrophic cardiomyopathy; however, they are also implicated in DCM.
- The most common genetic causes of DCM are truncating variants of the giant protein titin, encoded by TTN, which maintains sarcomere structure and acts as a key signaling molecule.
- Defects in the sarcolemmal membrane proteins are associated with DCM.
- The best known is dystrophin, encoded by the X chromosome gene DMD, abnormalities of which cause Duchenne's and Becker's muscle dystrophy.
- This protein provides a network that supports the sarcolemma and also connects to the sarcomere.
- The progressive functional defect in both cardiac and skeletal muscle reflects vulnerability to mechanical stress.
- Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias.
- Variants in SCN5A, the alpha subunit of the Nav 1.5 ion channel protein, distinct from those that cause the Brugada or long QT syndromes, have been implicated in DCM with conduction disease.
- Nuclear membrane protein defects in cardiac and skeletal muscle occur in either autosomal (lamin A/C) or X-linked (emerin) patterns.
- These defects are associated with a high prevalence of atrial and ventricular arrhythmias and conduction system disease, which can occur in some family members without or before detectable cardiomyopathy and underlie gene-specific risk stratification for sudden death and different utilization criteria for primary prevention implantable cardioverter-defibrillators in these patients.
- Intercalated disks contribute to intracellular connections, allowing mechanical and electrical coupling between cells and also connections to desmin filaments within the cell.
- Variants in proteins of the desmosomal complex compromise attachment of the myocytes, which can become disconnected and die via activation of Wnt/beta-catenin and proinflammatory signaling pathways, to be replaced by fat and fibrous tissue.
- These areas are highly arrhythmogenic and may dilate to form aneurysms.
- Although more often noted in the right ventricle (arrhythmogenic right ventricular cardiomyopathy), this condition can be restricted to the left ventricle (especially when secondary to truncating variants in DSP, which encodes desmoplakin) or affect both ventricles and has also been termed arrhythmogenic cardiomyopathy.
- Patients with an initial presentation of right ventricular cardiomyopathy who progress to include left ventricular dysfunction are at high risk for adverse events.
- Prognosis and therapy of DCM and ACM are dictated primarily by the stage of clinical disease and the risk for sudden death, which also varies based on disease gene (higher for patients with variants in DES, DSP, DSC2, DCG2, FLNC, LMNA, PKP2, PLN, RBM20, SCN5A, and TMEM43).
- The rate of progression of disease is also heritable, with marked variation based on disease gene observed (e.g., patients with TTN truncating variants frequently experience recovery with medical therapy).
- Medical therapy is generally guided by the phenotype, such that patients with DCM and reduced LVEF are treated with therapies recommended for heart failure with reduced ejection fraction.
- As for HCM, shared decision-making is needed regarding implantable cardioverter-defibrillators.
- However, the specific genotype plays a greater role in decisions regarding DCM, for which some features may warrant implantable cardioverter-defibrillator placement before the LVEF has declined to 0.35, such as the presence of pathogenic LMNA or FLNC variants or a family history of sudden death at early ages.
9. SPECIAL CONSIDERATIONS¶
- Vigorous physical activity and competitive sports have been historically prohibited; however, recent studies have failed to identify associated relationship between exertion and ventricular arrhythmias in hypertrophic cardiomyopathy, empowering patients and providers to make shared decisions about exercise.
- Women appear to have lower penetrance of sarcomere variants and an older age at hypertrophic cardiomyopathy diagnosis but subsequently increased rates of heart failure and mortality thereafter.
- In MYBPC3 variant carriers, the average age of disease development is ~40 years, while 30% remain free from hypertrophy after 70 years.
- Related individuals who carry the same variant may have a different extent and pattern of hypertrophy (e.g., asymmetric vs concentric), occurrence of outflow tract obstruction, and associated clinical outcomes.
- Sudden death and progression to heart failure occur more commonly in families with that history.
- Sex appears to play a role, as penetrance and clinical severity may be greater in men for most cardiomyopathies.
- The clinical course of a patient usually cannot be predicted based on which variant is present; thus, current therapy is based on the phenotype rather than the genetic defect.
- Currently, the greatest utility of genetic testing for cardiomyopathy is to inform family evaluations.
- However, genetic testing can provide risk stratification in some forms of cardiomyopathy and occasionally enables the detection of a disease for which specific therapy is indicated, such as the replacements for defective metabolic enzymes in Fabry's disease and Gaucher's disease.
- Moreover, clinical trials are interrogating the role of gene therapies for cardiomyopathy, which will provide greater impetus for genetic testing.
- For any patient with suspected or proven genetic disease, family members should be considered and evaluated in a longitudinal fashion.
- Screening generally includes both an echocardiogram and electrocardiogram (ECG).
- The indications and implications for confirmatory specific genetic testing vary depending on the specific variant.
- The identification of a disease-causing variant in a proband can focus family evaluations on variant carriers, but this strategy requires a high degree of certainty that the variant is truly pathogenic and not a benign DNA variant.
- Biopsy is not recommended to diagnose hypertrophic cardiomyopathy but can be used to exclude infiltrative and metabolic diseases.
- Rigorous athletic training (athlete's heart) may cause intermediate degrees of physiologic hypertrophy difficult to differentiate from mild hypertrophic cardiomyopathy.
- Unlike hypertrophic cardiomyopathy, hypertrophy in the athlete's heart regresses with cessation of training and is accompanied by supernormal exercise capacity (VO2max >50 mL/kg per min), mild ventricular dilation, and normal diastolic function.
- Most restrictive cardiomyopathy (RCM) is due to acquired causes, and there is increasing emphasis to diagnose amyloidosis due to transthyretin variants.
- Inherited metabolic and storage diseases can cause RCM, as can variants in DES causing combined cardiac and skeletal myopathy and sarcomere variants causing an overlap of RCM and hypertrophic cardiomyopathy.
- Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells.
- Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing pseudohypertrophy, usually with an abnormally short PR interval.
- The pseudohypertrophic phenotype is most common, but restrictive cardiomyopathy and DCM may occur.
- Most of these diseases are diagnosed during childhood.
- Fabry's disease results from a deficiency of the lysosomal enzyme alpha-galactosidase A caused by variants in GLA.
- This disorder of glycosphingolipid metabolism is an X-linked disorder that may also cause clinical disease in female carriers.
- Glycolipid accumulation may be limited to the cardiac tissues but usually also involves the skin, peripheral nerve, and kidney.
- Electron microscopy of endomyocardial biopsy tissue shows diagnostic vesicles containing concentric lamellar figures.
- Diagnosis can be made through assessment of enzyme activity and/or GLA.
- Gene therapy using a viral vector to deliver functional LAMP2 to cardiomyocytes is currently under study in humans with Danon disease.
10. KEY PEARLS & CLINICAL TRAPS¶
- Hypertrophic cardiomyopathy prevalence is approximately 1:500 in North America, Africa, and Asia.
- It is a leading cause of sudden death in the young and is an important cause of heart failure.
- Pediatric presentation is associated with increased early morbidity and mortality.
- Patients diagnosed as adults have decreased survival compared to age-matched individuals without hypertrophic cardiomyopathy.
- Women appear to have lower penetrance of sarcomere variants and an older age at hypertrophic cardiomyopathy diagnosis but subsequently increased rates of heart failure and mortality thereafter.
- In MYBPC3 variant carriers, the average age of disease development is ~40 years, while 30% remain free from hypertrophy after 70 years.
- Related individuals who carry the same variant may have a different extent and pattern of hypertrophy (e.g., asymmetric vs concentric), occurrence of outflow tract obstruction, and associated clinical outcomes.
- Sudden death and progression to heart failure occur more commonly in families with that history.
- Truncating variants in TTN, encoding the giant sarcomeric protein titin, are the most common cause of DCM, accounting for up to 25% of familial disease.
- On average, men with TTN truncating variants develop cardiomyopathy a decade before women, without distinctive clinical features.
- Variants in thick and thin filament genes account for ~8% of DCM and may manifest in early childhood.
- Familial clustering is present in ~30–40% of cases of DCM and arrhythmogenic cardiomyopathy (ACM).
- Monogenic etiologies can be identified in ~25% of DCM and ACM cases.
- Sarcomere variants are most associated with hypertrophic cardiomyopathy; however, they are also implicated in DCM.
- The most common genetic causes of DCM are truncating variants of the giant protein titin, encoded by TTN, which maintains sarcomere structure and acts as a key signaling molecule.
- Defects in the sarcolemmal membrane proteins are associated with DCM.
- The best known is dystrophin, encoded by the X chromosome gene DMD, abnormalities of which cause Duchenne's and Becker's muscle dystrophy.
- This protein provides a network that supports the sarcolemma and also connects to the sarcomere.
- The progressive functional defect in both cardiac and skeletal muscle reflects vulnerability to mechanical stress.
- Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias.
- Variants in SCN5A, the alpha subunit of the Nav 1.5 ion channel protein, distinct from those that cause the Brugada or long QT syndromes, have been implicated in DCM with conduction disease.
- Nuclear membrane protein defects in cardiac and skeletal muscle occur in either autosomal (lamin A/C) or X-linked (emerin) patterns.
- These defects are associated with a high prevalence of atrial and ventricular arrhythmias and conduction system disease, which can occur in some family members without or before detectable cardiomyopathy and underlie gene-specific risk stratification for sudden death and different utilization criteria for primary prevention implantable cardioverter-defibrillators in these patients.
- Intercalated disks contribute to intracellular connections, allowing mechanical and electrical coupling between cells and also connections to desmin filaments within the cell.
- Variants in proteins of the desmosomal complex compromise attachment of the myocytes, which can become disconnected and die via activation of Wnt/beta-catenin and proinflammatory signaling pathways, to be replaced by fat and fibrous tissue.
- These areas are highly arrhythmogenic and may dilate to form aneurysms.
- Although more often noted in the right ventricle (arrhythmogenic right ventricular cardiomyopathy), this condition can be restricted to the left ventricle (especially when secondary to truncating variants in DSP, which encodes desmoplakin) or affect both ventricles and has also been termed arrhythmogenic cardiomyopathy.
- Patients with an initial presentation of right ventricular cardiomyopathy who progress to include left ventricular dysfunction are at high risk for adverse events.
- Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells.
- Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing pseudohypertrophy, usually with an abnormally short PR interval.
- The pseudohypertrophic phenotype is most common, but restrictive cardiomyopathy and DCM may occur.
- Most of these diseases are diagnosed during childhood.
- Fabry's disease results from a deficiency of the lysosomal enzyme alpha-galactosidase A caused by variants in GLA.
- This disorder of glycosphingolipid metabolism is an X-linked disorder that may also cause clinical disease in female carriers.
- Glycolipid accumulation may be limited to the cardiac tissues but usually also involves the skin, peripheral nerve, and kidney.
- Electron microscopy of endomyocardial biopsy tissue shows diagnostic vesicles containing concentric lamellar figures.
- Diagnosis can be made through assessment of enzyme activity and/or GLA.
- Most restrictive cardiomyopathy (RCM) is due to acquired causes, and there is increasing emphasis to diagnose amyloidosis due to transthyretin variants.
- Inherited metabolic and storage diseases can cause RCM, as can variants in DES causing combined cardiac and skeletal myopathy and sarcomere variants causing an overlap of RCM and hypertrophic cardiomyopathy.
- Rigorous athletic training (athlete's heart) may cause intermediate degrees of physiologic hypertrophy difficult to differentiate from mild hypertrophic cardiomyopathy.
- Unlike hypertrophic cardiomyopathy, hypertrophy in the athlete's heart regresses with cessation of training and is accompanied by supernormal exercise capacity (VO2max >50 mL/kg per min), mild ventricular dilation, and normal diastolic function.
- Vigorous physical activity and competitive sports have been historically prohibited; however, recent studies have failed to identify associated relationship between exertion and ventricular arrhythmias in hypertrophic cardiomyopathy, empowering patients and providers to make shared decisions about exercise.
- Women appear to have lower penetrance of sarcomere variants and an older age at hypertrophic cardiomyopathy diagnosis but subsequently increased rates of heart failure and mortality thereafter.
- In MYBPC3 variant carriers, the average age of disease development is ~40 years, while 30% remain free from hypertrophy after 70 years.
- Related individuals who carry the same variant may have a different extent and pattern of hypertrophy (e.g., asymmetric vs concentric), occurrence of outflow tract obstruction, and associated clinical outcomes.
- Sudden death and progression to heart failure occur more commonly in families with that history.
- Sex appears to play a role, as penetrance and clinical severity may be greater in men for most cardiomyopathies.
- The clinical course of a patient usually cannot be predicted based on which variant is present; thus, current therapy is based on the phenotype rather than the genetic defect.
- Currently, the greatest utility of genetic testing for cardiomyopathy is to inform family evaluations.
- However, genetic testing can provide risk stratification in some forms of cardiomyopathy and occasionally enables the detection of a disease for which specific therapy is indicated, such as the replacements for defective metabolic enzymes in Fabry's disease and Gaucher's disease.
- Moreover, clinical trials are interrogating the role of gene therapies for cardiomyopathy, which will provide greater impetus for genetic testing.
- For any patient with suspected or proven genetic disease, family members should be considered and evaluated in a longitudinal fashion.
- Screening generally includes both an echocardiogram and electrocardiogram (ECG).
- The indications and implications for confirmatory specific genetic testing vary depending on the specific variant.
- The identification of a disease-causing variant in a proband can focus family evaluations on variant carriers, but this strategy requires a high degree of certainty that the variant is truly pathogenic and not a benign DNA variant.
- Biopsy is not recommended to diagnose hypertrophic cardiomyopathy but can be used to exclude infiltrative and metabolic diseases.
- Rigorous athletic training (athlete's heart) may cause intermediate degrees of physiologic hypertrophy difficult to differentiate from mild hypertrophic cardiomyopathy.
- Unlike hypertrophic cardiomyopathy, hypertrophy in the athlete's heart regresses with cessation of training and is accompanied by supernormal exercise capacity (VO2max >50 mL/kg per min), mild ventricular dilation, and normal diastolic function.
- Most restrictive cardiomyopathy (RCM) is due to acquired causes, and there is increasing emphasis to diagnose amyloidosis due to transthyretin variants.
- Inherited metabolic and storage diseases can cause RCM, as can variants in DES causing combined cardiac and skeletal myopathy and sarcomere variants causing an overlap of RCM and hypertrophic cardiomyopathy.
- Multiple genetic disorders of metabolic pathways can cause myocardial disease, due to infiltration of abnormal products or cells containing them between the myocytes, and storage disease, due to their accumulation within cells.
- Hypertrophic cardiomyopathy may be mimicked by the myocardium thickened with these abnormal products causing pseudohypertrophy, usually with an abnormally short PR interval.
- The pseudohypertrophic phenotype is most common, but restrictive cardiomyopathy and DCM may occur.
- Most of these diseases are diagnosed during childhood.
- Fabry's disease results from a deficiency of the lysosomal enzyme alpha-galactosidase A caused by variants in GLA.
- This disorder of glycosphingolipid metabolism is an X-linked disorder that may also cause clinical disease in female carriers.
- Glycolipid accumulation may be limited to the cardiac tissues but usually also involves the skin, peripheral nerve, and kidney.
- Electron microscopy of endomyocardial biopsy tissue shows diagnostic vesicles containing concentric lamellar figures.
- Diagnosis can be made through assessment of enzyme activity and/or GLA.
- Gene therapy using a viral vector to deliver functional LAMP2 to cardiomyocytes is currently under study in humans with Danon disease.
Flowcharts & Algorithms¶
Reproduced from Harrison's 22nd Edition.
Flowchart 1¶

Caption: FIGURE 267-5 Treatment algorithm for hypertrophic cardiomyopathy depending on the with obstruction to outflow. Note that all patients with hypertrophic cardiomyopathy require treatment for symptoms. ICD, implantable cardioverter-defibrillator; LV, left
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶
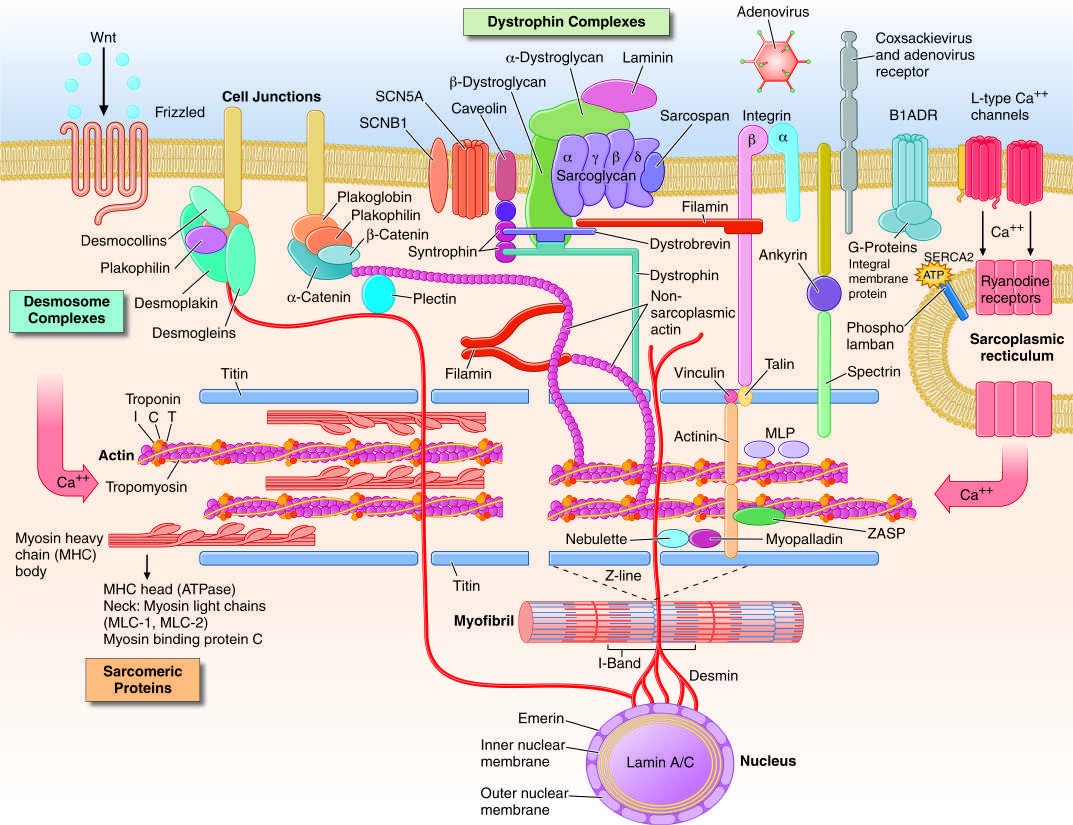
Caption: FIGURE 267-1 Drawing of myocyte indicating multiple sites of abnormal gene products proteins (actin, myosin, tropomyosin, and the associated regulatory proteins), the structures, the desmosome complexes associated with cell-cell connections and adenosine triphosphate. (Figure adapted from Jeffrey A. Towbin, MD, University of — Figure 267-1: Drawing of myocyte indicating multiple sites of abnormal gene products associated with cardiomyopathy, including sarcomeric proteins, dystrophin complex, desmosome complexes, and cytoskeletal proteins.
Figure 2¶
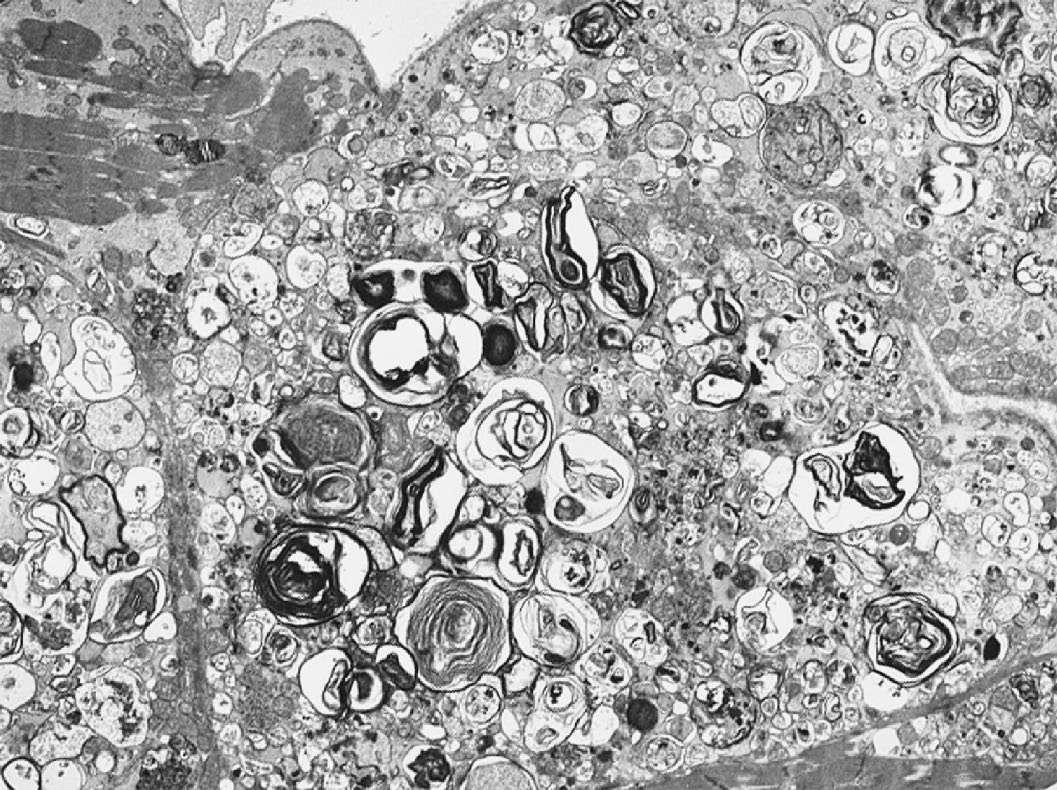
Caption: FIGURE 267-9 Fabry’s disease. Transmission electron micrograph of a right ventricular endomyocardial biopsy specimen at high magnification showing the characteristic concentric lamellar inclusions of glycosphingolipids accumulating as a result of deficiency of the lysosomal enzyme alpha-galactosidase A. Image taken at 15,000× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.) — Figure 267-2: Echocardiogram of hypertrophic cardiomyopathy showing asymmetric hypertrophy of the septum compared to the lateral wall of the left ventricle (LV) with anterior mitral valve motion.
Figure 3¶
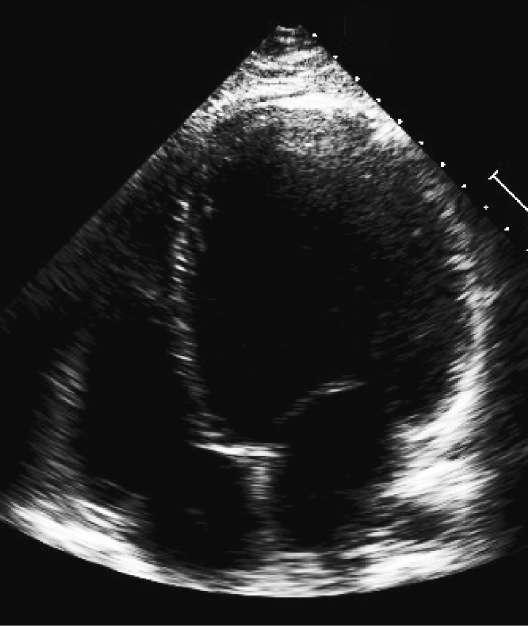
Caption: FIGURE 267-8 Dilated cardiomyopathy. Microscopic specimen of a dilated cardiomyopathy showing the nonspecific changes of interstitial fibrosis and myocyte hypertrophy characterized by increased myocyte size and enlarged, irregular nuclei. Hematoxylin and eosin–stained section, 100× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.) to mechanical stress. Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias, FIGURE 267-6 Dilated cardiomyopathy. This gross specimen of a heart removed at the time of transplantation shows massive left ventricular dilation and moderate but variants in SCN5A, the α subunit of the Nav 1.5 ion channel pro- — Figure 267-3: Gross specimen of a heart with hypertrophic cardiomyopathy removed at the time of transplantation, showing asymmetric septal hypertrophy with the septum bulging into the left ventricular outflow tract.
Figure 4¶
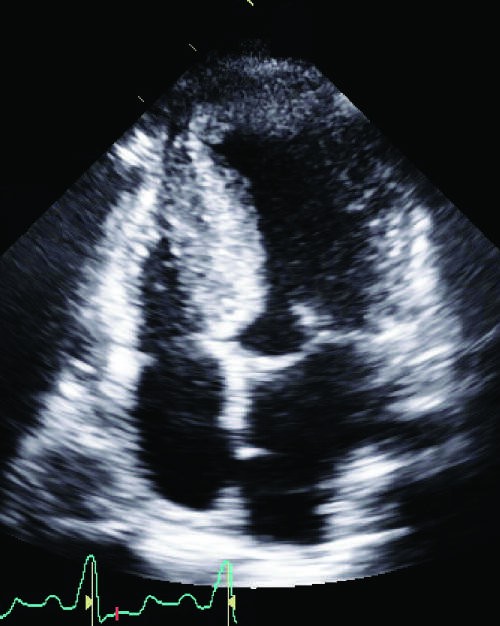
Caption: FIGURE 267-4 Hypertrophic cardiomyopathy. Microscopic image of hypertrophic LA cardiomyopathy showing the characteristic disarrayed myocyte architecture with swirling and branching rather than the usual parallel arrangement of myocyte fibers. Myocyte nuclei vary markedly in size, and interstitial fibrosis is present. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.) — Figure 267-4: Microscopic image of hypertrophic cardiomyopathy showing characteristic disarrayed myocyte architecture with swirling and branching fibers, variable nuclear size, and interstitial fibrosis.
Figure 5¶

Caption: FIGURE 267-8 Dilated cardiomyopathy. Microscopic specimen of a dilated cardiomyopathy showing the nonspecific changes of interstitial fibrosis and myocyte hypertrophy characterized by increased myocyte size and enlarged, irregular nuclei. Hematoxylin and eosin–stained section, 100× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.) to mechanical stress. Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias, FIGURE 267-6 Dilated cardiomyopathy. This gross specimen of a heart removed at the time of transplantation shows massive left ventricular dilation and moderate but variants in SCN5A, the α subunit of the Nav 1.5 ion channel pro- — Figure 267-5: Treatment algorithm for hypertrophic cardiomyopathy depending on the presence and severity of symptoms and the presence of an intraventricular gradient with obstruction to outflow.
Figure 6¶
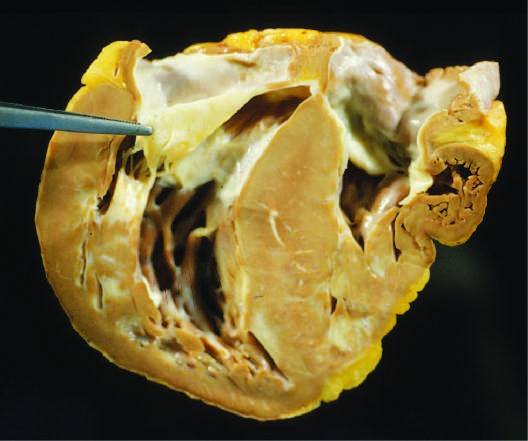
Caption: FIGURE 267-4 Hypertrophic cardiomyopathy. Microscopic image of hypertrophic LA cardiomyopathy showing the characteristic disarrayed myocyte architecture with swirling and branching rather than the usual parallel arrangement of myocyte fibers. Myocyte nuclei vary markedly in size, and interstitial fibrosis is present. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.) — Figure 267-6: Gross specimen of a heart removed at the time of transplantation showing massive left ventricular dilation and moderate right ventricular dilation with wall thinning.
Figure 7¶
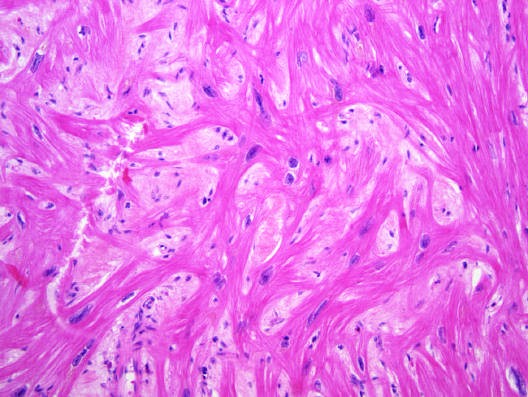
Caption: FIGURE 267-4 Hypertrophic cardiomyopathy. Microscopic image of hypertrophic LA cardiomyopathy showing the characteristic disarrayed myocyte architecture with swirling and branching rather than the usual parallel arrangement of myocyte fibers. Myocyte nuclei vary markedly in size, and interstitial fibrosis is present. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.) — Figure 267-7: Echocardiogram of a young man with dilated cardiomyopathy showing massive global dilation and thinning of the walls of the left ventricle (LV) with enlarged left atrium (LA).
Figure 8¶
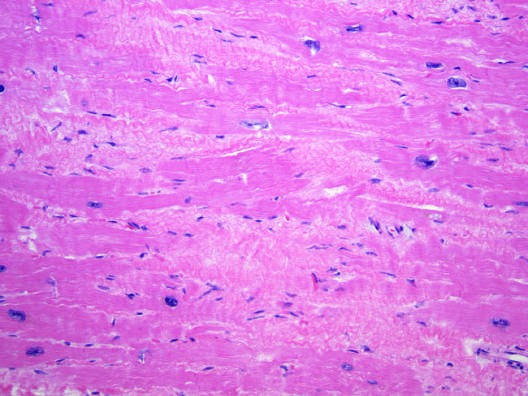
Caption: FIGURE 267-8 Dilated cardiomyopathy. Microscopic specimen of a dilated cardiomyopathy showing the nonspecific changes of interstitial fibrosis and myocyte hypertrophy characterized by increased myocyte size and enlarged, irregular nuclei. Hematoxylin and eosin–stained section, 100× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.) to mechanical stress. Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias, FIGURE 267-6 Dilated cardiomyopathy. This gross specimen of a heart removed at the time of transplantation shows massive left ventricular dilation and moderate but variants in SCN5A, the α subunit of the Nav 1.5 ion channel pro- — Figure 267-8: Microscopic specimen of a dilated cardiomyopathy showing nonspecific changes of interstitial fibrosis and myocyte hypertrophy characterized by increased myocyte size and enlarged, irregular nuclei.
Figure 9¶

Caption: Figure 267-9: Transmission electron micrograph of a right ventricular endomyocardial biopsy specimen showing characteristic concentric lamellar inclusions of glycosphingolipids accumulating as a result of deficiency of the lysosomal enzyme alpha-galactosidase A in Fabry's disease.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.