Histoplasmosis¶
Chapter 218 | Part 5: Infectious Diseases · Part 5 – Infectious Diseases: Fungal
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- Histoplasma capsulatum is a thermal dimorphic fungus endemic to the Ohio and Mississippi river valleys.
- Diagnosis relies on Histoplasma antigen detection (>95% sensitivity in progressive disseminated histoplasmosis), culture, and serology.
- Treatment for severe disease involves lipid Amphotericin B followed by Itraconazole; mild cases may resolve without therapy.
- Progressive disseminated histoplasmosis (PDH) occurs in immunocompromised hosts (e.g., AIDS, transplant) and requires prolonged antifungal therapy.
- Fibrosing mediastinitis is a rare but fatal complication characterized by progressive fibrosis around mediastinal structures.
- Posaconazole is an effective salvage therapy for refractory cases, including histoplasmosis, though controlled trials are lacking.
- Chronic cavitary histoplasmosis mimics tuberculosis and is seen in smokers with structural lung disease.
- Serologic tests (ID, CF, EIA) are useful for chronic pulmonary histoplasmosis but may persist for years after infection.
- Itraconazole requires therapeutic drug monitoring (target 2–5 μg/mL) due to P450 interactions and variable absorption.
- Unlike latent tuberculosis, inactive histoplasmosis does not reactivate in immunocompetent hosts.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Immune Response
- 3.2 Dissemination
- 4. CLINICAL FEATURES
- 4.1 Acute Pulmonary Histoplasmosis
- 4.2 Chronic Cavitary Histoplasmosis
- 4.3 Progressive Disseminated Histoplasmosis (PDH)
- 4.4 Fibrosing Mediastinitis
- 5. DIFFERENTIAL DIAGNOSIS
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Diagnostic Tests
- 6.2 Diagnostic Criteria & Recommendations
- 7. MANAGEMENT & TREATMENT
- 7.1 Drug Regimens
- 7.2 Monitoring & Follow-up
- 8. PROGNOSIS & COMPLICATIONS
- 9. SPECIAL CONSIDERATIONS
- 10. KEY PEARLS & CLINICAL TRAPS
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🖼 Figure | Spiked spherical conidia of H |
| 2 | 🖼 Figure | Intracellular yeasts (arrows) of H |
| 3 | 🖼 Figure | Spiked spherical conidia of H |
1. DEFINITION & OVERVIEW¶
- Histoplasmosis is an endemic mycosis caused by the thermal dimorphic fungus Histoplasma capsulatum.
- The disease spectrum ranges from asymptomatic infection to life-threatening disseminated illness.
- In most endemic areas in North America, H. capsulatum var. capsulatum is the causative agent.
- In Central and South America, histoplasmosis is common and is caused by genetically different clades of H. capsulatum var. capsulatum.
- In Africa, H. capsulatum var. duboisii is also found; yeasts of var. duboisii are larger than those of var. capsulatum.
- African histoplasmosis caused by var. duboisii is clinically distinct and is characterized by frequent skin and bone involvement.
- The clinical features and management of histoplasmosis caused by the genetically different clades in Central and South America are similar to those of the disease in North America.
2. EPIDEMIOLOGY¶
- Histoplasmosis is the most prevalent endemic mycosis in North America.
- Endemicity is particularly notable in the Ohio and Mississippi river valleys of North America.
- Endemic areas also include certain parts of Mexico, Central and South America (Brazil), Africa, and Asia.
- Histoplasmosis is increasingly reported outside of traditionally known endemic areas.
- The geographic distribution of histoplasmosis is related to the humid and acidic nature of the soil in the endemic areas.
- Soil enriched with bird or bat droppings promotes the growth and sporulation of Histoplasma.
- Disruption of soil containing the organism leads to aerosolization of the microconidia and exposure of humans nearby.
- Activities associated with high-level exposure and high infection attack rate include spelunking, excavation, cleaning of chicken coops, demolition and remodeling of old buildings, and cutting of dead trees.
- Most cases seen outside of highly endemic areas represent imported disease, e.g., in Europe, histoplasmosis is diagnosed fairly often, mostly in emigrants from or travelers to endemic areas on other continents.
- The epidemiology of histoplasmosis is changing as a result of global climate changes and with the continued expansion of at-risk populations and acceleration of intercontinental and international travel.
- The population at risk for histoplasmosis continues to grow as a result of increasing numbers of patients receiving immunosuppressive therapies for autoimmune disorders, cancers, and organ transplants.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- Histoplasma capsulatum, a thermal dimorphic fungus, is the etiologic agent of histoplasmosis.
- Mycelia—the naturally infectious form of Histoplasma—have a characteristic appearance, with microconidial and macroconidial forms.
- Microconidia are oval and are small enough (2–4 μm) to reach the terminal bronchioles and alveoli.
- Shortly after infecting the host, mycelia transform into the yeasts that are found inside macrophages and other phagocytes.
- The yeast forms are characteristically small (2–5 μm), with occasional narrow budding.
- In the laboratory, mycelia are best grown at room temperature, whereas yeasts are grown at 37°C on enriched media.
- Infection follows inhalation of microconidia.
- Once they reach the alveolar spaces, microconidia are rapidly recognized and engulfed by alveolar macrophages, where they transform into yeasts.
- This process is integral to the pathogenesis of histoplasmosis and is dependent on the availability of calcium and iron inside the phagocytes.
- The yeasts are capable of evading the immune system and multiplying inside resting macrophages.
- Neutrophils and then lymphocytes are attracted to the site of infection.
- Before the development of cellular immunity, yeasts use the phagosomes as a vehicle for translocation to local draining lymph nodes, whence they spread hematogenously throughout the reticuloendothelial system.
- Effective cellular immunity develops ~2 weeks after infection.
- T cells produce interferon-γ to assist the macrophages in killing the organism and controlling the progression of disease.
- Interleukin 12 and tumor necrosis factor α (TNF-α) play an essential role in cellular immunity to H. capsulatum.
- In the immunocompetent host, macrophages, lymphocytes, and epithelial cells eventually organize and form granulomas that contain the organisms.
- These granulomas typically fibrose and calcify; calcified lung nodules, mediastinal lymph nodes, and hepatosplenic calcifications are frequently found in healthy individuals from endemic areas.
- In immunocompetent hosts, infection with H. capsulatum confers protective immunity to reinfection.
- In patients with impaired cellular immunity, the infection is not properly contained and can disseminate throughout the reticuloendothelial system.
- Progressive disseminated histoplasmosis (PDH) can involve multiple organs, most commonly the lungs, bone marrow, spleen, liver, adrenal glands, and mucocutaneous membranes.
- Unlike latent tuberculosis, inactive histoplasmosis does not reactivate.
- In patients with mildly impaired immune systems, active infection may smolder and eventually worsen with further decline in immunity.
- Structural lung disease (e.g., emphysema) impairs the clearance of pulmonary histoplasmosis leading to the development of chronic pulmonary disease.
- This chronic process is characterized by progressive inflammation, tissue necrosis, and fibrosis mimicking cavitary tuberculosis.
3.1 Immune Response¶
- Effective cellular immunity develops ~2 weeks after infection.
- T cells produce interferon-γ to assist the macrophages in killing the organism and controlling the progression of disease.
- Interleukin 12 and tumor necrosis factor α (TNF-α) play an essential role in cellular immunity to H. capsulatum.
- In the immunocompetent host, macrophages, lymphocytes, and epithelial cells eventually organize and form granulomas that contain the organisms.
- These granulomas typically fibrose and calcify; calcified lung nodules, mediastinal lymph nodes, and hepatosplenic calcifications are frequently found in healthy individuals from endemic areas.
- In immunocompetent hosts, infection with H. capsulatum confers protective immunity to reinfection.
3.2 Dissemination¶
- In patients with impaired cellular immunity, the infection is not properly contained and can disseminate throughout the reticuloendothelial system.
- Progressive disseminated histoplasmosis (PDH) can involve multiple organs, most commonly the lungs, bone marrow, spleen, liver, adrenal glands, and mucocutaneous membranes.
- In patients with mildly impaired immune systems, active infection may smolder and eventually worsen with further decline in immunity.
- Structural lung disease (e.g., emphysema) impairs the clearance of pulmonary histoplasmosis leading to the development of chronic pulmonary disease.
- This chronic process is characterized by progressive inflammation, tissue necrosis, and fibrosis mimicking cavitary tuberculosis.
4. CLINICAL FEATURES¶
- The clinical spectrum of histoplasmosis ranges from asymptomatic infection to life-threatening illness.
- The attack rate and the extent and severity of the disease depend on the intensity of exposure, the immune status of the exposed individual, and the underlying lung architecture of the host.
- Common manifestations include fever, weight loss, hepatosplenomegaly, and thrombocytopenia.
- Other findings may include meningitis or focal brain lesions, ulcerations of the oral mucosa, gastrointestinal ulcerations and bleeding, and adrenal insufficiency.
- Prompt recognition of this devastating illness is of paramount importance in patients with severe manifestations or with underlying immunosuppression, especially those due to AIDS or anti-TNF therapy.
- Chronic cavitary histoplasmosis is seen in smokers who have structural lung disease (e.g., bullous emphysema).
- This chronic illness is characterized by productive cough, dyspnea, low-grade fever, night sweats, and weight loss.
- Chest radiographs usually show upper-lobe infiltrates, cavitation, and pleural thickening—findings resembling those of tuberculosis.
- Without treatment, the course is slowly progressive.
- Fibrosing mediastinitis is an uncommon but serious complication of histoplasmosis.
- In certain patients, acute infection is followed for unknown reasons by progressive fibrosis around the hilar and mediastinal lymph nodes, encasing mediastinal structures with potentially devastating consequences.
- Major manifestations include superior vena cava syndrome, obstruction of pulmonary vessels, and airway obstruction.
- Patients may experience recurrent pneumonia, hemoptysis, or respiratory failure.
- Fibrosing mediastinitis is fatal in up to one-third of cases.
- In healed histoplasmosis, calcified mediastinal nodes or lung parenchymal nodules may erode through the walls of the airways and cause hemoptysis and expectoration of calcified material.
- This condition is called broncholithiasis.
- In immunocompetent individuals with low-level exposure, most frequent Histoplasma infections are either asymptomatic or mild and self-limited.
- Of adults residing in endemic areas, up to 75% have immunologic and/or radiographic evidence of previous infection without clinical manifestations.
- Asymptomatic lung nodules representing controlled histoplasmosis are frequently found on chest computed tomography (CT) scans obtained during screening for lung cancer in smokers from endemic areas.
- When symptoms of acute histoplasmosis develop, they usually appear 1–4 weeks after exposure.
- Heavy exposure leads to a flulike illness with fever, chills, sweats, headache, myalgia, anorexia, dry cough, dyspnea, and chest pain.
- Chest radiographs usually show signs of pneumonitis with prominent hilar or mediastinal adenopathy.
- Pulmonary infiltrates may be focal with light exposure or diffuse with heavy exposure.
- Rheumatologic symptoms of arthralgia or arthritis, often associated with erythema nodosum, occur in 5–10% of patients with acute histoplasmosis.
- Pericarditis may also develop.
- These manifestations represent inflammatory responses to the acute pulmonary infection rather than extrapulmonary spread.
- Affected hilar or mediastinal lymph nodes may undergo necrosis and coalesce to form large mediastinal masses that can cause compression of great vessels, proximal airways, and the esophagus.
- These necrotic lymph nodes may also rupture and create fistulas between mediastinal structures (e.g., bronchoesophageal fistulae).
4.1 Acute Pulmonary Histoplasmosis¶
- Heavy exposure leads to a flulike illness with fever, chills, sweats, headache, myalgia, anorexia, dry cough, dyspnea, and chest pain.
- Chest radiographs usually show signs of pneumonitis with prominent hilar or mediastinal adenopathy.
- Pulmonary infiltrates may be focal with light exposure or diffuse with heavy exposure.
- Rheumatologic symptoms of arthralgia or arthritis, often associated with erythema nodosum, occur in 5–10% of patients with acute histoplasmosis.
- Pericarditis may also develop.
- These manifestations represent inflammatory responses to the acute pulmonary infection rather than extrapulmonary spread.
- Affected hilar or mediastinal lymph nodes may undergo necrosis and coalesce to form large mediastinal masses that can cause compression of great vessels, proximal airways, and the esophagus.
- These necrotic lymph nodes may also rupture and create fistulas between mediastinal structures (e.g., bronchoesophageal fistulae).
4.2 Chronic Cavitary Histoplasmosis¶
- Chronic cavitary histoplasmosis is seen in smokers who have structural lung disease (e.g., bullous emphysema).
- This chronic illness is characterized by productive cough, dyspnea, low-grade fever, night sweats, and weight loss.
- Chest radiographs usually show upper-lobe infiltrates, cavitation, and pleural thickening—findings resembling those of tuberculosis.
- Without treatment, the course is slowly progressive.
4.3 Progressive Disseminated Histoplasmosis (PDH)¶
- PDH is typically seen in immunocompromised individuals, who account for ~70% of cases.
- Common risk factors include AIDS (CD4+ T-cell count, <200/μL), extremes of age, the administration of immunosuppressive medications to prevent or treat rejection following transplantation (e.g., prednisone, mycophenolate, calcineurin inhibitors), and the use of methotrexate, anti-TNF-α agents, and other biologic response modifiers for autoimmune disorders.
- PDH may also occur in healthy individuals, some of whom may have rare undiagnosed genetic immunodeficiencies of the relevant immune pathways (IFN-γ and TNF-α).
- Workup for these conditions should be considered in healthy subjects with PDH.
- The clinical spectrum of PDH ranges from an acute, rapidly fatal course—with diffuse interstitial or reticulonodular lung infiltrates and difficulty breathing.
- In most other cases of pulmonary histoplasmosis, treatment is not recommended, especially if the immune system of the host is intact and the degree of exposure is not heavy.
- The symptoms usually are mild, subacute, and not progressive, and the illness resolves without therapy.
- Treatment should be considered if the symptoms are not improving within a month.
4.4 Fibrosing Mediastinitis¶
- Fibrosing mediastinitis is an uncommon but serious complication of histoplasmosis.
- In certain patients, acute infection is followed for unknown reasons by progressive fibrosis around the hilar and mediastinal lymph nodes, encasing mediastinal structures with potentially devastating consequences.
- Major manifestations include superior vena cava syndrome, obstruction of pulmonary vessels, and airway obstruction.
- Patients may experience recurrent pneumonia, hemoptysis, or respiratory failure.
- Fibrosing mediastinitis is fatal in up to one-third of cases.
5. DIFFERENTIAL DIAGNOSIS¶
- Chronic cavitary histoplasmosis findings resemble those of tuberculosis.
- Cross-reactivity occurs with African histoplasmosis, blastomycosis, coccidioidomycosis, paracoccidioidomycosis, talaromycosis, and rarely aspergillosis.
- Other fungal infections may present with similar pulmonary infiltrates and difficulty breathing.
- Malignancy (e.g., lymphoma, carcinoma) may present with mediastinal masses and constitutional symptoms.
- Other mycobacterial infections (e.g., tuberculosis) may present with cavitary lung disease and systemic symptoms.
6. INVESTIGATIONS & DIAGNOSIS¶
- Fungal culture remains the gold standard diagnostic test for histoplasmosis.
- However, culture results may not be known for up to 1 month, and cultures are often negative in less severe cases.
- Cultures are positive in ~75% of patients with PDH and chronic pulmonary histoplasmosis.
- Cultures of bronchoalveolar lavage (BAL) fluid are positive in about half of patients with acute pulmonary histoplasmosis causing diffuse infiltrates and hypoxemia.
- In PDH, the culture yield is highest for BAL fluid, bone marrow aspirate, and blood.
- Cultures of sputum or bronchial washings are usually positive in chronic pulmonary histoplasmosis.
- Cultures are typically negative, however, in other forms of histoplasmosis.
- Fungal stains of cytopathology or biopsy materials showing structures resembling Histoplasma yeasts are helpful in the diagnosis of PDH, yielding positive results in about half of cases.
- Yeasts can be seen in BAL fluid from patients with diffuse pulmonary infiltrates, in bone marrow biopsy samples, and in biopsy specimens of other involved organs (e.g., liver, adrenal glands).
- Occasionally, yeasts are seen within circulating phagocytes on blood smears from patients with severe PDH.
- However, staining artifacts and other fungal elements sometimes stain positively and may be misidentified as Histoplasma yeasts.
- Culture and pathology are no longer performed in most patients because diagnosis is more often established by antigen detection and/or serology, more rapidly and without subjecting the patient to invasive procedures.
- The detection of Histoplasma antigen in body fluids is extremely useful in the diagnosis of PDH and acute diffuse pulmonary histoplasmosis.
- The sensitivity of this method is >95% in patients with PDH and >80% in patients with severe acute pulmonary histoplasmosis resulting from heavy exposure, if both urine and serum are tested.
- Antigen levels correlate with severity of illness in PDH and can be used to follow disease progression, as levels predictably decrease with effective therapy.
- Increasing antigen levels also predict relapse.
- Histoplasma antigen can be detected in cerebrospinal fluid from patients with Histoplasma meningitis and in BAL fluid from those with pulmonary histoplasmosis.
- Lateral flow antigen detection allows the diagnosis of histoplasmosis at the bedside as a point-of-care testing method, which might offer access to rapid testing in resource-limited areas of the world where traditional antigen testing might not be available.
- Serologic tests, including immunodiffusion (ID), complement fixation (CF), and IgG and IgM enzyme immunoassay (EIA), are useful for the diagnosis of histoplasmosis, especially in immunocompetent patients.
- One month may be required for the detection of antibodies after the onset of infection by ID or CF, but antibodies may be detected earlier by more sensitive methods (EIA).
- IgM appears first then declines, and IgG appears later and increases during the infection.
- EIA for IgG and IgM antibodies provides a more accurate method for monitoring changes and antibody levels.
- Serologic tests are especially useful for the diagnosis of chronic pulmonary histoplasmosis.
- Limitations of ID and CF, however, include insensitivity early in the course of infection and reduced sensitivity in immunosuppressed patients, especially those receiving immunosuppression for organ transplantation.
- Also, antibodies may persist for several years after infection.
- Positive results from past infection may lead to a misdiagnosis of active histoplasmosis in a patient with another disease process.
- Recommendations for the diagnosis and treatment of histoplasmosis are summarized in Table 218-1.
6.1 Diagnostic Tests¶
- Fungal culture remains the gold standard diagnostic test for histoplasmosis.
- However, culture results may not be known for up to 1 month, and cultures are often negative in less severe cases.
- Cultures are positive in ~75% of patients with PDH and chronic pulmonary histoplasmosis.
- Cultures of bronchoalveolar lavage (BAL) fluid are positive in about half of patients with acute pulmonary histoplasmosis causing diffuse infiltrates and hypoxemia.
- In PDH, the culture yield is highest for BAL fluid, bone marrow aspirate, and blood.
- Cultures of sputum or bronchial washings are usually positive in chronic pulmonary histoplasmosis.
- Cultures are typically negative, however, in other forms of histoplasmosis.
- Fungal stains of cytopathology or biopsy materials showing structures resembling Histoplasma yeasts are helpful in the diagnosis of PDH, yielding positive results in about half of cases.
- Yeasts can be seen in BAL fluid from patients with diffuse pulmonary infiltrates, in bone marrow biopsy samples, and in biopsy specimens of other involved organs (e.g., liver, adrenal glands).
- Occasionally, yeasts are seen within circulating phagocytes on blood smears from patients with severe PDH.
- However, staining artifacts and other fungal elements sometimes stain positively and may be misidentified as Histoplasma yeasts.
- Culture and pathology are no longer performed in most patients because diagnosis is more often established by antigen detection and/or serology, more rapidly and without subjecting the patient to invasive procedures.
- The detection of Histoplasma antigen in body fluids is extremely useful in the diagnosis of PDH and acute diffuse pulmonary histoplasmosis.
- The sensitivity of this method is >95% in patients with PDH and >80% in patients with severe acute pulmonary histoplasmosis resulting from heavy exposure, if both urine and serum are tested.
- Antigen levels correlate with severity of illness in PDH and can be used to follow disease progression, as levels predictably decrease with effective therapy.
- Increasing antigen levels also predict relapse.
- Histoplasma antigen can be detected in cerebrospinal fluid from patients with Histoplasma meningitis and in BAL fluid from those with pulmonary histoplasmosis.
- Lateral flow antigen detection allows the diagnosis of histoplasmosis at the bedside as a point-of-care testing method, which might offer access to rapid testing in resource-limited areas of the world where traditional antigen testing might not be available.
- Serologic tests, including immunodiffusion (ID), complement fixation (CF), and IgG and IgM enzyme immunoassay (EIA), are useful for the diagnosis of histoplasmosis, especially in immunocompetent patients.
- One month may be required for the detection of antibodies after the onset of infection by ID or CF, but antibodies may be detected earlier by more sensitive methods (EIA).
- IgM appears first then declines, and IgG appears later and increases during the infection.
- EIA for IgG and IgM antibodies provides a more accurate method for monitoring changes and antibody levels.
- Serologic tests are especially useful for the diagnosis of chronic pulmonary histoplasmosis.
- Limitations of ID and CF, however, include insensitivity early in the course of infection and reduced sensitivity in immunosuppressed patients, especially those receiving immunosuppression for organ transplantation.
- Also, antibodies may persist for several years after infection.
- Positive results from past infection may lead to a misdiagnosis of active histoplasmosis in a patient with another disease process.
6.2 Diagnostic Criteria & Recommendations¶
- Recommendations for the diagnosis and treatment of histoplasmosis are summarized in Table 218-1.
- Once suspected, the diagnosis of histoplasmosis is usually straightforward as many diagnostic tools are now available in the United States.
- This is not the case in resource-limited endemic regions of Central America, South America, and Africa, where the diagnosis is often delayed, with consequently poor outcomes.
Table 1 — Table 218-1 Recommendations for the Diagnosis and Treatment of Histoplasmosis¶
| TYPE OF HISTOPLASMOSIS | DIAGNOSTIC TESTS | TREATMENT RECOMMENDATIONS | COMMENTS |
|---|---|---|---|
| Acute pulmonary, mild to moderate with no improvement by the time of diagnosis | Histoplasma antigen (BAL fluid, serum, urine) Cytopathology and fungal culture of BAL fluid Histoplasma serology (ID and CF), (EIA): IgG and IgM |
Itraconazole (200 mg bid) for 6–12 weeks. Monitor renal and hepatic function. |
Patients with mild cases usually recover without therapy, but itraconazole should be considered if the patient's condition is not already improving by the time the diagnosis is established. |
| Acute pulmonary, severe illness, respiratory failure (ARDS) | Histoplasma antigen (BAL fluid, serum, urine) Cytopathology and fungal culture of BAL fluid Histoplasma serology (ID and CF), (EIA): IgG and IgM |
Lipid AmB (3–5 mg/kg per day) ± glucocorticoids for 1–2 weeks; then itraconazole (200 mg bid) for 6–12 weeks. Monitor renal and hepatic function. |
|
| Chronic/cavitary pulmonary | Histoplasma serology (ID and CF), (EIA): IgG and IgM Fungal culture of sputum or BAL fluid |
Itraconazole (200 mg bid) Adjust dose to achieve blood levels of 2–5 μg/mL for at least 12 months. Monitor hepatic function. |
Continue treatment until radiographic findings show no further improvement. Monitor for relapse after treatment is stopped. |
| Progressive disseminated | Histoplasma antigen (BAL fluid, serum, urine) Histoplasma serology (ID and CF), (EIA): IgG and IgM Fungal culture of blood or bone marrow aspirate Cytopathology on biopsy of affected organ |
Lipid AmB (3–5 mg/kg per day) for 1–2 weeks; then itraconazole (200 mg bid); adjust dose to achieve blood levels of 2–5 μg/mL for at least 12 months. Monitor renal and hepatic function. |
|
| Central nervous system | Histoplasma antigen CSF Histoplasma serology (ID and CF), (EIA): IgG and IgM Fungal culture of CSF |
Liposomal AmB (5 mg/kg per day) for 4–6 weeks; then itraconazole (200 mg bid) Adjust dose to achieve blood levels of 2–5 μg/mL for at least 12 months. Monitor renal and hepatic function. |
A longer course of lipid AmB is recommended because of the high risk of relapse. Itraconazole should be continued until CSF or MRI abnormalities clear. |
7. MANAGEMENT & TREATMENT¶
- Treatment is indicated for all patients with PDH or chronic pulmonary histoplasmosis.
- Treatment is also indicated for most symptomatic patients with acute pulmonary histoplasmosis who have not improved by the time the diagnosis is established, especially in those with diffuse infiltrates and difficulty breathing.
- In most other cases of pulmonary histoplasmosis, treatment is not recommended, especially if the immune system of the host is intact and the degree of exposure is not heavy.
- The symptoms usually are mild, subacute, and not progressive, and the illness resolves without therapy.
- Treatment should be considered if the symptoms are not improving within a month.
- The preferred treatments for histoplasmosis include the lipid formulations of amphotericin B in severe cases and itraconazole in others.
- Liposomal amphotericin B is more effective and better tolerated than the deoxycholate formulation and is more effective in patients with AIDS and PDH.
- The deoxycholate formulation of amphotericin B is an alternative to a lipid formulation for patients at low risk for nephrotoxicity and if liposomal amphotericin B is not available.
- Posaconazole and isavuconazole are alternatives for patients who cannot take itraconazole.
- Histoplasma may develop resistance to fluconazole and voriconazole, and they are not the preferred alternative to itraconazole, especially in immunocompromised patients.
- In severe cases requiring hospitalization, a lipid formulation of amphotericin B is used first for 2 weeks, followed by itraconazole.
- In patients with meningitis, a lipid formulation of amphotericin B should be given for 4–6 weeks before switching to itraconazole.
- In immunosuppressed patients, the degree of immunosuppression should be reduced if possible, although immune reconstitution inflammatory syndrome (IRIS) may ensue.
- Antiretroviral treatment improves the outcome of PDH in patients with AIDS and is recommended; however, whether antiretroviral treatment should be delayed to avoid IRIS is unknown.
- Blood levels of itraconazole should be monitored to ensure adequate drug exposure, with target concentrations of the parent drug and its hydroxy metabolites measuring 2–5 μg/mL.
- Drug interactions should be carefully assessed; itraconazole not only is cleared by cytochrome P450 metabolism but also inhibits cytochrome P450.
- This profile causes interactions with many other medications used routinely in organ transplant recipients.
- The duration of treatment for acute pulmonary histoplasmosis is 6–12 weeks.
- The duration of treatment for PDH and chronic pulmonary histoplasmosis is at least 1 year.
- Antigen levels in urine and serum should be monitored during and for at least 1 year after therapy.
- Posaconazole has been reported to be effective salvage therapy for aspergillosis, mucormycosis, fusariosis, cryptococcosis, histoplasmosis, and coccidioidomycosis, although controlled clinical trials are lacking.
- The tablet formulation is not hampered by the suboptimal absorption that occurs with the suspension; the tablet also results in higher and more reliable blood levels of the drug.
- Posaconazole is less hepatotoxic than voriconazole and does not cause the skin, visual, or bone toxicity that occurs with voriconazole.
- However, the use of posaconazole is linked to significant P450-related drug interactions and can lead to pseudohyperaldosteronism, which may manifest with hypokalemia, edema, and/or hypertension.
7.1 Drug Regimens¶
- Acute pulmonary, mild to moderate with no improvement by the time of diagnosis: Itraconazole (200 mg bid) for 6–12 weeks. Monitor renal and hepatic function.
- Acute pulmonary, severe illness, respiratory failure (ARDS): Lipid AmB (3–5 mg/kg per day) ± glucocorticoids for 1–2 weeks; then itraconazole (200 mg bid) for 6–12 weeks. Monitor renal and hepatic function.
- Chronic/cavitary pulmonary: Itraconazole (200 mg bid) Adjust dose to achieve blood levels of 2–5 μg/mL for at least 12 months. Monitor hepatic function.
- Progressive disseminated: Lipid AmB (3–5 mg/kg per day) for 1–2 weeks; then itraconazole (200 mg bid); adjust dose to achieve blood levels of 2–5 μg/mL for at least 12 months. Monitor renal and hepatic function.
- Central nervous system: Liposomal AmB (5 mg/kg per day) for 4–6 weeks; then itraconazole (200 mg bid) Adjust dose to achieve blood levels of 2–5 μg/mL for at least 12 months. Monitor renal and hepatic function.
- Posaconazole: Effective salvage therapy for aspergillosis, mucormycosis, fusariosis, cryptococcosis, histoplasmosis, and coccidioidomycosis. Tablet formulation results in higher and more reliable blood levels. Less hepatotoxic than voriconazole. Linked to significant P450-related drug interactions and can lead to pseudohyperaldosteronism (hypokalemia, edema, hypertension).
- Itraconazole: Target concentrations of the parent drug and its hydroxy metabolites measuring 2–5 μg/mL. Drug interactions should be carefully assessed; itraconazole is cleared by cytochrome P450 metabolism but also inhibits cytochrome P450. This profile causes interactions with many other medications used routinely in organ transplant recipients.
7.2 Monitoring & Follow-up¶
- Antigen levels in urine and serum should be monitored during and for at least 1 year after therapy.
- Increasing antigen levels also predict relapse.
- Histoplasma antigen can be detected in cerebrospinal fluid from patients with Histoplasma meningitis and in BAL fluid from those with pulmonary histoplasmosis.
- Lateral flow antigen detection allows the diagnosis of histoplasmosis at the bedside as a point-of-care testing method, which might offer access to rapid testing in resource-limited areas of the world where traditional antigen testing might not be available.
- Continue treatment until radiographic findings show no further improvement. Monitor for relapse after treatment is stopped.
8. PROGNOSIS & COMPLICATIONS¶
- Fibrosing mediastinitis is fatal in up to one-third of cases.
- In immunosuppressed patients, the degree of immunosuppression should be reduced if possible, although immune reconstitution inflammatory syndrome (IRIS) may ensue.
- Antiretroviral treatment improves the outcome of PDH in patients with AIDS and is recommended; however, whether antiretroviral treatment should be delayed to avoid IRIS is unknown.
- Without treatment, the course of chronic cavitary histoplasmosis is slowly progressive.
- In immunocompetent individuals with low-level exposure, most frequent Histoplasma infections are either asymptomatic or mild and self-limited.
- Of adults residing in endemic areas, up to 75% have immunologic and/or radiographic evidence of previous infection without clinical manifestations.
- Asymptomatic lung nodules representing controlled histoplasmosis are frequently found on chest computed tomography (CT) scans obtained during screening for lung cancer in smokers from endemic areas.
9. SPECIAL CONSIDERATIONS¶
- In resource-limited endemic regions of Central America, South America, and Africa, the diagnosis is often delayed, with consequently poor outcomes.
- In immunosuppressed patients, the degree of immunosuppression should be reduced if possible, although immune reconstitution inflammatory syndrome (IRIS) may ensue.
- Antiretroviral treatment improves the outcome of PDH in patients with AIDS and is recommended; however, whether antiretroviral treatment should be delayed to avoid IRIS is unknown.
- Blood levels of itraconazole should be monitored to ensure adequate drug exposure, with target concentrations of the parent drug and its hydroxy metabolites measuring 2–5 μg/mL.
- Drug interactions should be carefully assessed; itraconazole not only is cleared by cytochrome P450 metabolism but also inhibits cytochrome P450.
- This profile causes interactions with many other medications used routinely in organ transplant recipients.
- The duration of treatment for acute pulmonary histoplasmosis is 6–12 weeks, while that for PDH and chronic pulmonary histoplasmosis is at least 1 year.
- Posaconazole and isavuconazole are alternatives for patients who cannot take itraconazole.
- Histoplasma may develop resistance to fluconazole and voriconazole, and they are not the preferred alternative to itraconazole, especially in immunocompromised patients.
10. KEY PEARLS & CLINICAL TRAPS¶
- Unlike latent tuberculosis, inactive histoplasmosis does not reactivate.
- Cross-reactivity occurs with African histoplasmosis, blastomycosis, coccidioidomycosis, paracoccidioidomycosis, talaromycosis, and rarely aspergillosis.
- Fibrosing mediastinitis is fatal in up to one-third of cases.
- In immunocompetent individuals with low-level exposure, most frequent Histoplasma infections are either asymptomatic or mild and self-limited.
- Of adults residing in endemic areas, up to 75% have immunologic and/or radiographic evidence of previous infection without clinical manifestations.
- Asymptomatic lung nodules representing controlled histoplasmosis are frequently found on chest computed tomography (CT) scans obtained during screening for lung cancer in smokers from endemic areas.
- Treatment should be considered if the symptoms are not improving within a month.
- Liposomal amphotericin B is more effective and better tolerated than the deoxycholate formulation and is more effective in patients with AIDS and PDH.
- The deoxycholate formulation of amphotericin B is an alternative to a lipid formulation for patients at low risk for nephrotoxicity and if liposomal amphotericin B is not available.
- Posaconazole has been reported to be effective salvage therapy for aspergillosis, mucormycosis, fusariosis, cryptococcosis, histoplasmosis, and coccidioidomycosis, although controlled clinical trials are lacking.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶
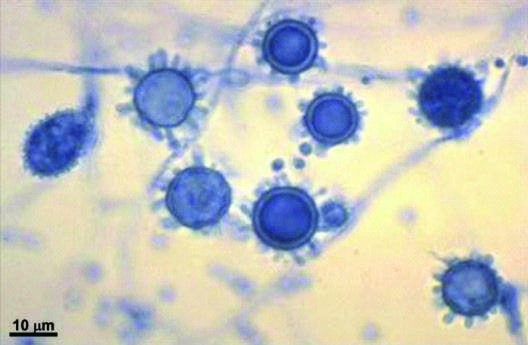
Caption: FIGURE 218-1 Spiked spherical conidia of H. capsulatum (lacto-phenol cotton blue stain) grown in the laboratory at room temperature. — Spiked spherical conidia of H. capsulatum (lacto-phenol cotton blue stain) grown in the laboratory at room temperature, showing microconidial and macroconidial forms.
Figure 2¶

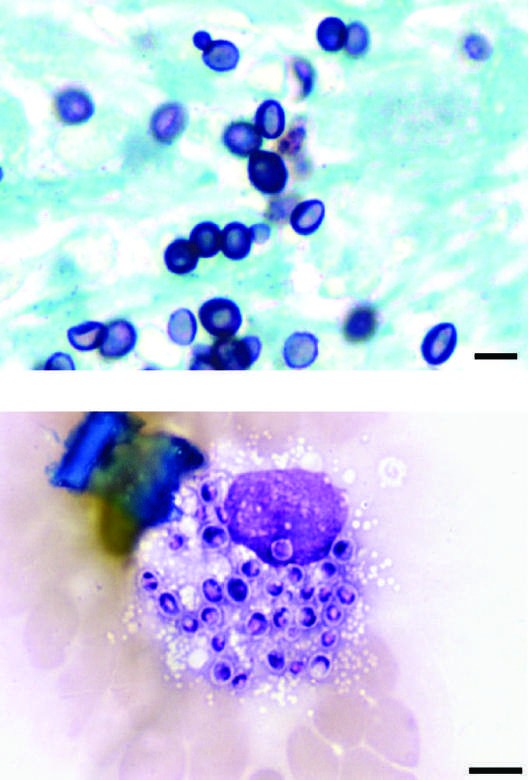
Caption: FIGURE 218-3 Intracellular yeasts (arrows) of H. capsulatum in a liver biopsy specimen (hematoxylin and eosin stain) from a patient who developed progressive disseminated histoplasmosis while receiving anti–tumor necrosis factor therapy for rheumatoid arthritis. — Small (2–5 μm) narrow budding yeasts of H. capsulatum from bronchoalveolar lavage fluid (Grocott's methenamine silver stain) and intracellular yeasts within an alveolar macrophage (Giemsa stain).
Figure 3¶
Caption: FIGURE 218-1 Spiked spherical conidia of H. capsulatum (lacto-phenol cotton blue stain) grown in the laboratory at room temperature. — Intracellular yeasts (arrows) of H. capsulatum in a liver biopsy specimen (hematoxylin and eosin stain) from a patient who developed progressive disseminated histoplasmosis while receiving anti–tumor necrosis factor therapy.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.