Inflammatory Myopathies¶
Chapter 377 | Part 11: Immune-Mediated, Inflammatory, and Rheumatologic Disorders · Part 11 – Rheumatology & Immunology
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- Inflammatory myopathies (IMs) include dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM), antisynthetase syndrome (ASyS), and inclusion body myositis (IBM).
- DM is associated with malignancy (15% within first 2-3 years in adults) and specific autoantibodies (anti-MDA5, anti-TIF1-γ, anti-NXP2).
- IBM is the most common cause of myopathy in adults >50 years, characterized by asymmetric weakness of finger/wrist flexors and quadriceps, and poor response to immunotherapy.
- IMNM is associated with anti-HMGCR or anti-SRP antibodies and can be triggered by statin use; anti-HMGCR myopathy does not improve with statin discontinuation.
- ASyS is defined by myositis, nonerosive arthritis, ILD, Raynaud's, mechanic hands, and fever with anti-aminoacyl-tRNA synthetase antibodies (e.g., anti-Jo-1).
- CK is the most sensitive marker of muscle destruction; >2000 U/L is almost always due to myopathy.
- Muscle biopsy is required to distinguish IMs, especially to exclude IBM in suspected PM cases.
- IBM is not typically responsive to immunotherapy; treatment focuses on physical/occupational therapy and swallowing management.
- Malignancy screening is crucial in DM, PM, and IMNM, especially with anti-TIF1-γ or anti-NXP2 antibodies.
- ILD is associated with anti-MDA5 DM and ASyS; spiral chest CT is best for honeycomb pattern.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Dermatomyositis Pathogenesis
- 3.2 Inclusion Body Myositis Pathogenesis
- 3.3 Immune-Mediated Necrotizing Myopathy Pathogenesis
- 3.4 Antisynthetase Syndrome Pathogenesis
- 4. CLINICAL FEATURES
- 4.1 Dermatomyositis Clinical Features
- 4.2 Polymyositis Clinical Features
- 4.3 Immune-Mediated Necrotizing Myopathy Clinical Features
- 4.4 Antisynthetase Syndrome Clinical Features
- 4.5 Inclusion Body Myositis Clinical Features
- 5. DIFFERENTIAL DIAGNOSIS
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Laboratory Features
- 6.2 Imaging and Biopsy
- 7. MANAGEMENT & TREATMENT
- 7.1 Glucocorticoids
- 7.2 Immunotherapies
- 8. PROGNOSIS & COMPLICATIONS
- 9. SPECIAL CONSIDERATIONS
- 10. KEY PEARLS & CLINICAL TRAPS
- Figures & Illustrations
📋 Figures in This Chapter¶
1. DEFINITION & OVERVIEW¶
- Inflammatory myopathies (IMs) are a group of disorders characterized by muscle inflammation and weakness.
- Major types include dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM), antisynthetase syndrome (ASyS), and inclusion body myositis (IBM).
- Other IMs include those caused by infection, eosinophilic myositis, granulomatous myositis, and myositis triggered by checkpoint inhibitors.
- Inflammatory cell infiltrates can occasionally be seen in muscle biopsies of hereditary myopathies (e.g., muscular dystrophies, metabolic myopathies), sporadic late-onset nemaline myopathy (SLONM), and toxic myopathies.
- Epidemiologic studies suggest the incidence of IM grouped together is up to 16 cases per 100,000 with prevalence in the range of 14–32 per 100,000.
- Defining the actual incidence and prevalence is limited by different diagnostic criteria employed in various epidemiologic studies.
- Increasing recognition of ASyS and IMNM, as well as frequent misdiagnosis of IBM, complicates epidemiology.
- Idiopathic PM without signs of an overlap syndrome is quite rare compared to DM, ASyS, IBM, and IMNM that occur in roughly similar frequencies.
- DM can occur in children (juvenile DM), while IBM always occurs in adults and is the most common cause of myopathy in those aged >50.
- DM, PM, and ASyS are more common in women, while IBM is more common in men.
2. EPIDEMIOLOGY¶
- Incidence of IM grouped together is up to 16 cases per 100,000.
- Prevalence is in the range of 14–32 per 100,000.
- DM can occur in children (juvenile DM) or adults.
- IBM always occurs in adults and is the most common cause of myopathy in those aged >50.
- DM, PM, and ASyS are more common in women.
- IBM is more common in men.
- Prevalence of gastrointestinal involvement in Behçet syndrome changes significantly across different populations (up to 50% in the Far East but rare in the Middle East).
- Patient characteristics, such as being young and male, need to be kept in mind when making treatment decisions as these patients tend to have a worse prognosis.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- Pathogenesis of DM was traditionally attributed to an antibody-mediated attack on endothelial cells, followed by complement-mediated destruction of capillaries and watershed ischemia of muscle fibers.
- Subsequent studies suggest this is not likely the case; immunoglobulin deposition is largely absent on endothelial cells.
- Complement deposition may be a secondary phenomenon.
- Increasing evidence suggests microvasculopathy and skin and muscle damage associated with DM are primarily due to toxicity from type I interferon (IFN)–mediated pathways, most likely IFN-β.
- MSAs are directly pathogenic; for example, anti-Mi-2 antibodies appear to be capable of entering myonuclei and inhibiting the CHD4/NuRD complex in the nucleosome.
- Pathogenesis of IBM is poorly understood; prominent adaptive immune system abnormalities related to T-cell inflammation and presence of a relatively specific autoantibody against a muscle protein indicate an autoimmune attack on muscle.
- Chronic and highly inflammatory environment within muscles in IBM may alter protein synthesis and degradation pathways in part via aberrant immunoproteasome expression.
- Additional histologic features, typically referred to as 'degenerative,' include aggregation of various proteins including markers of endoplasmic reticulum (ER) stress and autophagy (e.g., p62 and LC3).
- TDP-43, which is important for normal splicing of messenger RNA, is extruded from myonuclei in IBM; loss of TDP-43-mediated splicing repression likely leads to abnormal inclusion of cryptic exons in skeletal muscle and aberrant translation of muscle proteins.
- Pathogenesis of IMNM is not completely understood and likely varies depending on subtype.
- A trial of a complement inhibitor in both anti-HMGCR and anti-SRP myopathies failed to demonstrate any efficacy, so IMNM in these subtypes does not appear to be primarily complement driven.
- Recent studies suggest HMGCR antibodies may bind to the receptor on the sarcolemma and lead to accumulation of acetyl-CoA and subsequently an increase in lipids within muscle fibers.
- Recent studies also suggest anti-SRP antibodies are directly causal to the associated IMNM.
- ASyS is driven by gamma-IFN than type 1 IFN.
- Various antibodies may be directly pathogenic by binding to specific aminoacyl-tRNA synthetases thereby impairing protein synthesis.
3.1 Dermatomyositis Pathogenesis¶
- Microvasculopathy and skin and muscle damage associated with DM are primarily due to toxicity from type I interferon (IFN)–mediated pathways, most likely IFN-β.
- Immunoglobulin deposition is largely absent on endothelial cells.
- Complement deposition may be a secondary phenomenon.
- MSAs are directly pathogenic; for example, anti-Mi-2 antibodies appear to be capable of entering myonuclei and inhibiting the CHD4/NuRD complex in the nucleosome.
3.2 Inclusion Body Myositis Pathogenesis¶
- Prominent adaptive immune system abnormalities related to T-cell inflammation and presence of a relatively specific autoantibody against a muscle protein indicate an autoimmune attack on muscle.
- Chronic and highly inflammatory environment within muscles in IBM may alter protein synthesis and degradation pathways in part via aberrant immunoproteasome expression.
- Additional histologic features, typically referred to as 'degenerative,' include aggregation of various proteins including markers of endoplasmic reticulum (ER) stress and autophagy (e.g., p62 and LC3).
- TDP-43, which is important for normal splicing of messenger RNA, is extruded from myonuclei in IBM; loss of TDP-43-mediated splicing repression likely leads to abnormal inclusion of cryptic exons in skeletal muscle and aberrant translation of muscle proteins.
- Whether this tissue damage results directly from a pathogenic immune response or a secondary neurodegenerative process is unclear at this time.
3.3 Immune-Mediated Necrotizing Myopathy Pathogenesis¶
- Pathogenesis of IMNM is not completely understood and likely varies depending on subtype.
- A trial of a complement inhibitor in both anti-HMGCR and anti-SRP myopathies failed to demonstrate any efficacy, so IMNM in these subtypes does not appear to be primarily complement driven.
- Recent studies suggest HMGCR antibodies may bind to the receptor on the sarcolemma and lead to accumulation of acetyl-CoA and subsequently an increase in lipids within muscle fibers.
- Recent studies also suggest anti-SRP antibodies are directly causal to the associated IMNM.
3.4 Antisynthetase Syndrome Pathogenesis¶
- ASyS is driven by gamma-IFN than type 1 IFN.
- Various antibodies may be directly pathogenic by binding to specific aminoacyl-tRNA synthetases thereby impairing protein synthesis.
4. CLINICAL FEATURES¶
- DM manifests with symmetric, proximal greater than distal weakness along with a characteristic rash.
- Rash includes heliotrope rash (erythematous discoloration of eyelids with periorbital edema), Gottron sign (erythematous rash over the extensor surfaces of joints such as the knuckles, elbows, knees, and ankles), Gottron papules (raised erythematous rash over knuckles), V-sign (rash on the sun-exposed anterior neck and chest), shawl sign over the back of the neck and shoulders, nail bed telangiectasias, and subcutaneous calcium deposits.
- Weakness and rash usually accompany one another but can be separated by several months.
- Beyond 'classic DM' with prominent muscle and skin manifestations, there is a spectrum of involvement such that some patients have skin-predominant disease (only with a rash called amyopathic DM), or mini-mal muscle disease called hypomyopathic DM.
- Others may present mainly with weakness and little or no visible skin changes.
- Patients may also have myalgias, arthralgias, dysphagia, and dysarthria.
- Cutaneous disease activity is highly relevant in DM; in comparison to other debilitating skin diseases including cutaneous lupus erythematosus, psoriasis, and atopic dermatitis, skin symptoms in DM patients are associated with an overall reduction in life quality.
- Pruritus can be especially debilitating.
- Dyspnea can occur from ventilatory muscle weakness or intrinsic lung involvement including interstitial lung disease (ILD), bronchopneumonia, and alveolitis.
- Pulmonary manifestations are often associated with anti-MDA-5 antibodies or with antisynthetase antibodies; myositis associated with the ASyS is now considered a distinct disorder.
- DM can present in children (juvenile DM) or in adults.
- There is a higher risk for malignancy in adult-onset cases, ~15% within the first 2–3 years.
- PM usually presents with symmetric and proximal weakness that worsens over several weeks to months.
- As with DM, there can be associated heart, lung, and joint involvement as well as an increased risk of cancer.
- Some epidemiologic studies suggest that the risk of cancer in PM is less than that in DM, but these older series likely included patients with IBM and dystrophies with inflammation who were misdiagnosed as having PM.
- IMNM is characterized by the acute or insidious onset of symmetric, proximal more than distal weakness.
- Dysphagia, dysarthria, or myalgia may occur.
- Patients may have an underlying CTD (usually scleroderma or MCTD) or cancer (paraneoplastic necrotizing myopathy), or the condition may be idiopathic.
- There are at least two distinct forms of IMNM associated with specific autoantibodies (anti-HMGCR and anti-SRP).
- Anti-HMGCR myopathy can be seen in patients receiving statins, inhibitors of HMGCR, particularly in those aged >50 years.
- However, anti-HMGCR myopathy can develop in children and young adults without a history of statin use and can mimic a limb girdle muscular dystrophy.
- Unlike the more common 'toxic' myopathy associated with statin use, anti-HMGCR myopathy does not improve when statins are discontinued.
- Anti-SRP myopathies are notable for the presence of anti-SRP antibodies and a typically subacute, aggressive, and relatively refractory course.
- ASyS is defined by the presence of myositis, nonerosive arthritis, ILD, Raynaud's phenomenon, mechanic hands, and fever associated with antibodies against aminoacyl-tRNA synthetase.
- Some patients have an erythematous rash, and muscle biopsies share histopathologic features of DM, which likely accounts for many of these patients being classified as having DM.
- IBM usually manifests in patients over the age of 50 years and is slightly more common in men than women.
- It is associated with slowly progressive weakness and muscle atrophy that has a predilection for early involvement of the wrist and finger flexors in the arms and quadriceps in the legs.
- Weakness is often asymmetric.
- Dysphagia is common and rarely can be the presenting feature.
- These clinical features can help distinguish IBM from PM and other forms of myopathy.
- The mean duration from onset of symptoms to use of wheelchair or scooter is ~15 years.
- There is no known increase in risk of malignancy in IBM.
4.1 Dermatomyositis Clinical Features¶
- Symmetric, proximal greater than distal weakness along with a characteristic rash.
- Rash includes heliotrope rash (erythematous discoloration of eyelids with periorbital edema).
- Gottron sign (erythematous rash over the extensor surfaces of joints such as the knuckles, elbows, knees, and ankles).
- Gottron papules (raised erythematous rash over knuckles).
- V-sign (rash on the sun-exposed anterior neck and chest).
- Shawl sign over the back of the neck and shoulders.
- Nail bed telangiectasias.
- Subcutaneous calcium deposits.
- Weakness and rash usually accompany one another but can be separated by several months.
- Beyond 'classic DM' with prominent muscle and skin manifestations, there is a spectrum of involvement such that some patients have skin-predominant disease (only with a rash called amyopathic DM), or mini-mal muscle disease called hypomyopathic DM.
- Others may present mainly with weakness and little or no visible skin changes.
- Patients may also have myalgias, arthralgias, dysphagia, and dysarthria.
- Cutaneous disease activity is highly relevant in DM; in comparison to other debilitating skin diseases including cutaneous lupus erythematosus, psoriasis, and atopic dermatitis, skin symptoms in DM patients are associated with an overall reduction in life quality.
- Pruritus can be especially debilitating.
- Dyspnea can occur from ventilatory muscle weakness or intrinsic lung involvement including interstitial lung disease (ILD), bronchopneumonia, and alveolitis.
- Pulmonary manifestations are often associated with anti-MDA-5 antibodies or with antisynthetase antibodies; myositis associated with the ASyS is now considered a distinct disorder.
- DM can present in children (juvenile DM) or in adults.
- There is a higher risk for malignancy in adult-onset cases, ~15% within the first 2–3 years.
4.2 Polymyositis Clinical Features¶
- Usually presents with symmetric and proximal weakness that worsens over several weeks to months.
- As with DM, there can be associated heart, lung, and joint involvement as well as an increased risk of cancer.
- Some epidemiologic studies suggest that the risk of cancer in PM is less than that in DM, but these older series likely included patients with IBM and dystrophies with inflammation who were misdiagnosed as having PM.
4.3 Immune-Mediated Necrotizing Myopathy Clinical Features¶
- Characterized by the acute or insidious onset of symmetric, proximal more than distal weakness.
- Dysphagia, dysarthria, or myalgia may occur.
- Patients may have an underlying CTD (usually scleroderma or MCTD) or cancer (paraneoplastic necrotizing myopathy), or the condition may be idiopathic.
- There are at least two distinct forms of IMNM associated with specific autoantibodies (anti-HMGCR and anti-SRP).
- Anti-HMGCR myopathy can be seen in patients receiving statins, inhibitors of HMGCR, particularly in those aged >50 years.
- However, anti-HMGCR myopathy can develop in children and young adults without a history of statin use and can mimic a limb girdle muscular dystrophy.
- Unlike the more common 'toxic' myopathy associated with statin use, anti-HMGCR myopathy does not improve when statins are discontinued.
- Anti-SRP myopathies are notable for the presence of anti-SRP antibodies and a typically subacute, aggressive, and relatively refractory course.
4.4 Antisynthetase Syndrome Clinical Features¶
- Defined by the presence of myositis, nonerosive arthritis, ILD, Raynaud's phenomenon, mechanic hands, and fever associated with antibodies against aminoacyl-tRNA synthetase.
- Some patients have an erythematous rash, and muscle biopsies share histopathologic features of DM, which likely accounts for many of these patients being classified as having DM.
4.5 Inclusion Body Myositis Clinical Features¶
- Usually manifests in patients over the age of 50 years and is slightly more common in men than women.
- Associated with slowly progressive weakness and muscle atrophy that has a predilection for early involvement of the wrist and finger flexors in the arms and quadriceps in the legs.
- Weakness is often asymmetric.
- Dysphagia is common and rarely can be the presenting feature.
- These clinical features can help distinguish IBM from PM and other forms of myopathy.
- The mean duration from onset of symptoms to use of wheelchair or scooter is ~15 years.
- There is no known increase in risk of malignancy in IBM.
5. DIFFERENTIAL DIAGNOSIS¶
- In any patient presenting with weakness, the first step is to localize the site of the lesion by history and clinical findings.
- Weakness could be caused by a process in the cerebral hemispheres, spinal cord, anterior horn cell, peripheral nerve, neuromuscular junction, or muscle disorders.
- Past medical history, medication use, and family history, combined with a detailed clinical examination and an appreciation for the pattern of muscle involvement (e.g., what muscles are weak and atrophic or hypertrophic as well as the presence of scapular winging, early contractures, sensory abnormalities, fasciculations, or rash), help differentiate myopathies from other neuromuscular disorders and the different types of myopathies from each other.
- For example, atrophy with fasciculations suggests a neurogenic process such as amyotrophic lateral sclerosis.
- Fatigable weakness on examination points to a neuromuscular junction defect such as myasthenia gravis.
- Concomitant sensory symptoms suggest a central process such as a spinal cord disorder or a polyneuropathy.
- Scapular winging, calf hypertrophy or atrophy, and early contractures before significant weakness develops would strongly suggest a muscular dystrophy, particularly if there is a positive family history.
- A heliotrope rash combined with Gottron papules and dilated nailfold capillaries is diagnostic for DM.
- The presence of atrophy and weakness of the flexor forearm muscles and quadriceps in a person aged >50 years is most likely IBM.
- When the site of the lesion cannot be localized based on history and clinical examination alone, laboratory testing is required.
- Serum creatine kinase (CK) is the most sensitive laboratory marker of muscle destruction.
- Not all myopathies are associated with elevated CK levels, but a markedly elevated CK (e.g., >2000 U/L) is almost always due to a myopathy.
- A slightly elevated CK can also be seen in neurogenic processes.
- Fibromyalgia is likely in patients with a normal laboratory workup.
- In general, a muscle biopsy is not indicated unless there is objective weakness, an abnormal EMG, or elevated CK.
- Polymyalgia rheumatica should be considered in older individuals with an elevated erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) but normal CK and EMG.
6. INVESTIGATIONS & DIAGNOSIS¶
- Serum creatine kinase (CK) is the most sensitive laboratory marker of muscle destruction.
- Not all myopathies are associated with elevated CK levels, but a markedly elevated CK (e.g., >2000 U/L) is almost always due to a myopathy.
- A slightly elevated CK can also be seen in neurogenic processes.
- Serum aldolase may be increased in 10% of those with normal CK.
- Antinuclear antibodies can be positive but are a nonspecific finding.
- The myositis-specific antibodies (MSAs) that are specific for DM include anti–complex nucleosome remodeling histone deacetylase (anti-Mi-2), anti–transcription intermediary factor 1-γ (anti-TIF1-γ), anti–melanoma differentiation-associated gene 5 (anti-MDA5), anti–nuclear matrix protein 2 (anti-NXP-2), and anti–small ubiquitin-like modifier activating enzyme (anti-SAE).
- These antibodies are usually associated with characteristic clinical features, and recent studies suggest that they are also directly involved in the pathogenesis of DM.
- Mi-2 antibodies are found in 15–20% of patients with DM and are typically associated with an acute onset, a florid rash, and prominent weakness but a good response to therapy and a favorable prognosis.
- Anti-MDA5 antibodies are found in 10–20% of DM patients and up to 65% of patients with clinically amyopathic DM.
- This antibody is associated with palmar rash, severe skin ulcerations from ischemia, and rapidly progressive ILD.
- Anti-TIF1-γ, also known as p155, anti-bodies are found in adult cancer-associated DM with an 89% specificity and 70% sensitivity.
- Thus, enhanced vigilance for underlying cancer is especially important in these patients.
- Anti-NXP-2 antibodies are found in as many as 17% of patients with DM and are also associated with calcinosis, subcutaneous edema, distal weakness, and dysphagia, as well as with cancer.
- Anti-SAE antibodies are present in 1.5–8% of DM and are associated with an underlying cancer in 14–57% of patients.
- Most manifest with a skin rash alone, and CK is often normal, but approximately one-third of patients have elevated aldolase levels.
- ILD can also be seen in anti-SAE DM, but unlike anti-MDA5 amyopathic DM, the ILD is usually mild.
- Electromyography (EMG) and nerve conductions studies (NCS) are useful in localizing the site of the lesion but are less specific in helping to determine the actual cause of a myopathy.
- EMG can be useful at times in guiding what muscle to biopsy, especially if muscles typically biopsied are normal on clinical examination.
- Imaging skeletal muscle can be helpful in assessing muscle involvement and revealing fatty replacement, atrophy, or edema within muscle or surrounding fascia.
- A muscle biopsy is often required to definitively distinguish one myopathy from another, if there is no characteristic dermatomyositis rash or myositis specific autoantibody.
- However, a muscle biopsy should be performed in every case of suspected PM to exclude IBM (if not clinically apparent) and other causes of myopathy.
- Diagnosis of IMNM is by definition based upon histologic findings but again is not needed if a patient has clinical features and anti-3-hydroxy-3-methylglutaryl-coenzyme reductase (HMGCR) or anti–signal recognition particle (SRP) antibodies.
- It is important to biopsy a muscle that is clinically affected but not too weak (e.g., Medical Research Council grade 4 out of 5 in strength); otherwise, one may just see end-stage muscle.
- A biopsy should always be coordinated with an experienced muscle histopathology laboratory.
- Patients with severe muscle pain, subjective weakness, and fatigue with normal strength and function on examination are not likely to have an IM.
- Table 377-1 summarizes the clinical and laboratory features of inflammatory myopathies.
6.1 Laboratory Features¶
- Serum creatine kinase (CK) is the most sensitive laboratory marker of muscle destruction.
- Not all myopathies are associated with elevated CK levels, but a markedly elevated CK (e.g., >2000 U/L) is almost always due to a myopathy.
- A slightly elevated CK can also be seen in neurogenic processes.
- Serum aldolase may be increased in 10% of those with normal CK.
- Antinuclear antibodies can be positive but are a nonspecific finding.
- The myositis-specific antibodies (MSAs) that are specific for DM include anti–complex nucleosome remodeling histone deacetylase (anti-Mi-2), anti–transcription intermediary factor 1-γ (anti-TIF1-γ), anti–melanoma differentiation-associated gene 5 (anti-MDA5), anti–nuclear matrix protein 2 (anti-NXP-2), and anti–small ubiquitin-like modifier activating enzyme (anti-SAE).
- These antibodies are usually associated with characteristic clinical features, and recent studies suggest that they are also directly involved in the pathogenesis of DM.
- Mi-2 antibodies are found in 15–20% of patients with DM and are typically associated with an acute onset, a florid rash, and prominent weakness but a good response to therapy and a favorable prognosis.
- Anti-MDA5 antibodies are found in 10–20% of DM patients and up to 65% of patients with clinically amyopathic DM.
- This antibody is associated with palmar rash, severe skin ulcerations from ischemia, and rapidly progressive ILD.
- Anti-TIF1-γ, also known as p155, anti-bodies are found in adult cancer-associated DM with an 89% specificity and 70% sensitivity.
- Thus, enhanced vigilance for underlying cancer is especially important in these patients.
- Anti-NXP-2 antibodies are found in as many as 17% of patients with DM and are also associated with calcinosis, subcutaneous edema, distal weakness, and dysphagia, as well as with cancer.
- Anti-SAE antibodies are present in 1.5–8% of DM and are associated with an underlying cancer in 14–57% of patients.
- Most manifest with a skin rash alone, and CK is often normal, but approximately one-third of patients have elevated aldolase levels.
- ILD can also be seen in anti-SAE DM, but unlike anti-MDA5 amyopathic DM, the ILD is usually mild.
Table 1 — Table 377-1 Inflammatory Myopathies: Clinical and Laboratory Features¶
| DISORDER | SEX | AGE OF ONSET | RASH | PATTERN OF WEAKNESS | LABORATORY FEATURES | MUSCLE BIOPSY | CELLULAR INFILTRATE | RESPONSE TO IS THERAPY | COMMON ASSOCIATED CONDITIONS |
|---|---|---|---|---|---|---|---|---|---|
| DM | F > M | Childhood and adult | Yes | Proximal > distal | Normal or increased CK (up to 50× normal or higher); various MSAs (anti-MDA5, anti-TIF1, anti-Mi-2, anti-NXP2) | Perimysial and perivascular inflammation; IFN-1 regulated proteins (MHC-1, MxA), MAC deposition on capillaries | CD4+ dendritic cells; B cells; macrophages | Yes | Myocarditis, ILD, malignancy, vasculitis, other CTDs |
| PM | F > M | Adult | No | Proximal > distal | Increased CK (up to 50× normal or higher) | Endomysial and perivascular inflammation; ubiquitous expression of MHC-1 | CD8+ T cells; macrophages; plasma cells | Yes | |
| IMNM | M = F | Children and adults | No | Proximal > distal | Elevated CK (>10× normal or higher); anti-HMGCR or anti-SRP antibodies | Necrotic muscle fibers; minimal inflammatory infiltrate; MHC-I and MAC deposition on sarcolemma of scattered nonnecrotic muscle fibers | Macrophages in necrotic fibers undergoing phagocytosis | Yes | Malignancy, CTD, HMGCR antibody cases can be triggered by statin use |
| ASyS | F > M | Children and adults | Sometimes | Proximal > distal | Elevated CK (>10× normal or higher); antisynthetase antibodies | Perimysial and perivascular inflammation; perimysial fragmentation with alkaline phosphatase staining; perimysial muscle damage with necrosis; MHC-I, HLA-DR, and MAC deposition on sarcolemma of perifascicular muscle fibers | CD4+ dendritic cells; B cells; macrophages | Yes | |
| IBM | M > F | Older adults (>50 years) | No | Proximal and distal; predilection for: finger/wrist flexors, knee extensors | Normal or mildly increased CK (usually <10× normal); anti-cN-1A antibodies; large granular lymphocytes on flow cytometry and reduced CD4/CD8 ratio with increased CD8 count | Endomysial and perivascular inflammation; ubiquitous expression of MHC-1 and HLA-DR; rimmed vacuoles; p62, LC3, TDP-43 aggregates; EM: 15–18 nm tubulofilaments; ragged red and COX-negative fibers | CD8+ T cells; macrophages; plasma cells; myeloid dendritic cells; large granular lymphocytes | None or minimal | Granular lymphocytic leukemia/lymphocytosis, sarcoidosis, sicca or Sjögren's syndrome |
6.2 Imaging and Biopsy¶
- Electromyography (EMG) and nerve conductions studies (NCS) are useful in localizing the site of the lesion but are less specific in helping to determine the actual cause of a myopathy.
- EMG can be useful at times in guiding what muscle to biopsy, especially if muscles typically biopsied are normal on clinical examination.
- Imaging skeletal muscle can be helpful in assessing muscle involvement and revealing fatty replacement, atrophy, or edema within muscle or surrounding fascia.
- A muscle biopsy is often required to definitively distinguish one myopathy from another, if there is no characteristic dermatomyositis rash or myositis specific autoantibody.
- However, a muscle biopsy should be performed in every case of suspected PM to exclude IBM (if not clinically apparent) and other causes of myopathy.
- Diagnosis of IMNM is by definition based upon histologic findings but again is not needed if a patient has clinical features and anti-3-hydroxy-3-methylglutaryl-coenzyme reductase (HMGCR) or anti–signal recognition particle (SRP) antibodies.
- It is important to biopsy a muscle that is clinically affected but not too weak (e.g., Medical Research Council grade 4 out of 5 in strength); otherwise, one may just see end-stage muscle.
- A biopsy should always be coordinated with an experienced muscle histopathology laboratory.
- Patients with severe muscle pain, subjective weakness, and fatigue with normal strength and function on examination are not likely to have an IM.
- Polymyalgia rheumatica should be considered in older individuals with an elevated erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) but normal CK and EMG.
7. MANAGEMENT & TREATMENT¶
- Treatment is guided by type and severity of involvement, with the goal of preventing long-term damage.
- Most new manifestations tend to present within the first 5 years, and for most patients, the natural course is one of diminishing symptoms culminating in potential remission, frequently not requiring ongoing treatment with medications.
- Patient characteristics, such as being young and male, need to be kept in mind when making treatment decisions, as these patients tend to have a worse prognosis.
- In most patients, tapering and/or stopping their medications in 2–3 years after the symptoms have improved should be attempted.
- Oral ulcers can be managed with topical glucocorticoids and on an as-needed basis if mild.
- Lesions resistant to local measures may require systemic treatment with colchicine, oral glucocorticoids, immunosuppressants such as apremilast, azathioprine, or a tumor necrosis factor (TNF)-α inhibitor such as infliximab.
- A similar treatment approach can be used for genital ulcers and other mucocutaneous manifestations.
- Patients may need a combination of medications, at least initially, to control disease activity.
- Eye involvement, given its frequency and potential morbidity, requires early and aggressive treatment with brief courses of glucocorticoids and longer-term treatment with an immunosuppressant.
- Azathioprine is usually the preferred agent in clinical practice.
- TNF inhibitors infliximab or adalimumab can also be used, either as first-line monotherapy or more commonly in combination with systemic glucocorticoids and azathioprine, for control of disease activity.
- Cyclosporine can also be considered in combination regimens; monotherapy with interferon is another option.
- High-dose glucocorticoids are considered the first-line treatment.
- There is uncertainty regarding when to start second-line agents (e.g., methotrexate, azathioprine, mycophenolate, immunoglobulin, or rituximab).
- The clinician must weigh with the patient the increased risks of immunosuppression versus possible benefits (e.g., faster improvement, steroid-sparing effects, and/or avoidance of morbidities associated with long-term glucocorticoid use).
- It is general practice to start a second-line agent (typically methotrexate) with glucocorticoids in patients with severe weakness or other organ system involvement (e.g., myocarditis, ILD), those with increased risk of steroid complications (e.g., diabetics, osteoporosis, or postmenopausal women), and patients with IMNM who are known to have difficult-to-treat myositis.
- When treatment is initiated with prednisone alone, a second-line agent is added in patients who fail to significantly improve after 2–4 months of treatment or in those who cannot be tapered to a low dose of prednisone.
- Many patients with IMNM do not respond to prednisone alone or even prednisone plus a second-line agent in combination.
- Many require triple therapy with prednisone, methotrexate, and IVIG and, if this fails, rituximab.
- In our experience and that of others, anti-HMGCR myopathy often responds to monotherapy with IVIG.
- Unfortunately, IBM does not typically respond to any known immunotherapy.
- The mainstay of treatment is physical and occupational therapy to improve function, and swallowing therapy (and sometimes esophageal dilation or cricopharyngeal myotomy) in those with dysphagia.
- Table 377-2 summarizes immunotherapies for inflammatory myopathies.
7.1 Glucocorticoids¶
- Treatment is initiated with prednisone (0.75–1.5 mg/kg up to 100 mg) administered as a daily morning single dose (the most common dose used in adults is 60 mg daily).
- In patients with severe weakness or comorbidities (e.g., ILD, myocarditis), treatment with a short course of intravenous methylprednisolone (1 g daily for 3 days) is recommended prior to starting oral glucocorticoids.
- Patients are generally maintained on high-dose prednisone until strength normalizes or until improvement in strength has reached a plateau (usually 3–6 months).
- Subsequently, prednisone can be tapered by 5 mg every 2–4 weeks.
- Once the dose is reduced to 20 mg every day or every other day, the taper is slowed to 2.5 mg every 2–4 weeks.
- The goal is to taper prednisone.
7.2 Immunotherapies¶
- High-dose glucocorticoids are considered the first-line treatment.
- There is uncertainty regarding when to start second-line agents (e.g., methotrexate, azathioprine, mycophenolate, immunoglobulin, or rituximab).
- The clinician must weigh with the patient the increased risks of immunosuppression versus possible benefits (e.g., faster improvement, steroid-sparing effects, and/or avoidance of morbidities associated with long-term glucocorticoid use).
- It is general practice to start a second-line agent (typically methotrexate) with glucocorticoids in patients with severe weakness or other organ system involvement (e.g., myocarditis, ILD), those with increased risk of steroid complications (e.g., diabetics, osteoporosis, or postmenopausal women), and patients with IMNM who are known to have difficult-to-treat myositis.
- When treatment is initiated with prednisone alone, a second-line agent is added in patients who fail to significantly improve after 2–4 months of treatment or in those who cannot be tapered to a low dose of prednisone.
- Many patients with IMNM do not respond to prednisone alone or even prednisone plus a second-line agent in combination.
- Many require triple therapy with prednisone, methotrexate, and IVIG and, if this fails, rituximab.
- In our experience and that of others, anti-HMGCR myopathy often responds to monotherapy with IVIG.
- Unfortunately, IBM does not typically respond to any known immunotherapy.
- The mainstay of treatment is physical and occupational therapy to improve function, and swallowing therapy (and sometimes esophageal dilation or cricopharyngeal myotomy) in those with dysphagia.
- Table 377-2 summarizes immunotherapies for inflammatory myopathies.
Table 2 — Table 377-2 Immunotherapies for Inflammatory Myopathies¶
| THERAPY | ROUTE | DOSE | SIDE EFFECTS | MONITOR |
|---|---|---|---|---|
| Prednisone | Oral | 0.75–1.5 mg/kg per day to start | Hypertension, fluid and weight gain, hyperglycemia, hypokalemia, cataracts, gastric irritation, osteoporosis, infection, aseptic femoral necrosis | Weight, blood pressure, serum glucose/potassium, cataract formation |
| Methylprednisolone | Intravenous | 1 g in 100 mL/normal saline over 1–2 h, daily or every other day for 3–6 doses | Arrhythmia, flushing, dysgeusia, anxiety, insomnia, fluid and weight gain, hyperglycemia, hypokalemia, infection | Heart rate, blood pressure, serum glucose/potassium |
| Azathioprine | Oral | 2–3 mg/kg per day; single a.m. dose | Flu-like illness, hepatotoxicity, pancreatitis, leukopenia, macrocytosis, neoplasia, infection, teratogenicity | Blood count, liver enzymes |
| Methotrexate | Oral | 7.5–20 mg weekly, single or divided doses; 1 day a week dosing | Hepatotoxicity, pulmonary fibrosis, infection, neoplasia, infertility, leukopenia, alopecia, gastric irritation, stomatitis, teratogenicity | Liver enzymes, blood count |
| Methotrexate | Subcutaneously | 20–50 mg weekly; 1 day a week dosing | Same as oral | Same as oral |
| Cyclophosphamide | Oral | 1.5–2 mg/kg per day; single a.m. dose | Bone marrow suppression, infertility, hemorrhagic cystitis, alopecia, infections, neoplasia, teratogenicity | Blood count, urinalysis |
| Cyclophosphamide | Intravenous | 0.5–1.0 g/m2 per month × 6–12 months | Bone marrow suppression, infertility, hemorrhagic cystitis, alopecia, infections, neoplasia, teratogenicity | Blood count, urinalysis |
| Cyclosporine | Oral | 4–6 mg/kg per day, split into two daily doses | Nephrotoxicity, hypertension, infection, hepatotoxicity, hirsutism, tremor, gum hyperplasia, teratogenicity | Blood pressure, creatinine/BUN, liver enzymes, cyclosporine levels |
| Tacrolimus | Oral | 0.1–0.2 mg/kg per day in two divided doses | Nephrotoxicity, hypertension, infection, hepatotoxicity, hirsutism, tremor, gum hyperplasia, teratogenicity | Blood pressure, creatinine/BUN, liver enzymes, tacrolimus levels |
| Mycophenolate | Oral | Adults (1–1.5 g BID); Children (600 mg/m2 per dose BID); no >1 g/d in patients with renal failure | Bone marrow suppression, hypertension, tremor, diarrhea, nausea, vomiting, headache, sinusitis, confusion, amblyopia, cough, teratogenicity, infection, neoplasia | Blood count |
| Intravenous immunoglobulin | Intravenous | 2 g/kg over 2–5 days; then 1 g/kg every 4–8 weeks as needed | Hypotension, arrhythmia, diaphoresis, flushing, nephrotoxicity, headache, aseptic meningitis, anaphylaxis, stroke | Heart rate, blood pressure, aseptic creatinine/BUN |
| Rituximab | Intravenous | A course is typically 750 mg/m2 (up to 1 g) and repeated in 2 weeks; Courses are then repeated usually every 6–18 months | Infusion reactions (as per IVIG), infection, progressive multifocal leukoencephalopathy | Some check B-cell count prior to subsequent courses (but this may not be warranted) |
8. PROGNOSIS & COMPLICATIONS¶
- In the absence of malignancy, prognosis is generally favorable in patients with DM, with 5-year survival rates ranging from 70 to 93%.
- Poor prognostic features are increased age, associated ILD, cardiac disease, and late or previous inadequate treatment.
- Most patients with PM improve with immunotherapies but usually require lifelong treatment.
- Some retrospective studies suggest that PM does not respond as well as DM to these therapies.
- However, many of these older series of 'PM' likely included patients who actually had IMNM, IBM, or other myopathies (including muscular dystrophies) that do not respond to immunotherapies.
- As in DM, poor prognostic features are cancer, increased age, lung or cardiac involvement, and late or previously inadequate treatment.
- Anti-HMGCR myopathy is often responsive to intravenous immunoglobulin (IVIG) monotherapy.
- However, anti-SRP disease is difficult to treat, in particular for patients with interstitial lung disease.
- Aggressive treatment, often with early rituximab, is warranted in such cases.
- There does not appear to be an increased risk of malignancy in ASyS.
- Most patients respond to treatment, although ASyS can be difficult to treat, in particular for patients with interstitial lung disease.
- The myopathy is slowly progressive and is not typically responsive to immunotherapies.
- Most patients require a scooter or wheelchair within 10–15 years of onset of symptoms.
- IBM is slowly progressive and is not typically responsive to immunotherapies.
- Most patients require a scooter or wheelchair within 10–15 years of onset of symptoms.
9. SPECIAL CONSIDERATIONS¶
- DM can occur in children (juvenile DM), while IBM always occurs in adults and is the most common cause of myopathy in those aged >50.
- DM, PM, and ASyS are more common in women, while IBM is more common in men.
- Patient characteristics, such as being young and male, need to be kept in mind when making treatment decisions, as these patients tend to have a worse prognosis.
- In most patients, tapering and/or stopping their medications in 2–3 years after the symptoms have improved should be attempted.
- The clinician must weigh with the patient the increased risks of immunosuppression versus possible benefits (e.g., faster improvement, steroid-sparing effects, and/or avoidance of morbidities associated with long-term glucocorticoid use).
- It is general practice to start a second-line agent (typically methotrexate) with glucocorticoids in patients with severe weakness or other organ system involvement (e.g., myocarditis, ILD), those with increased risk of steroid complications (e.g., diabetics, osteoporosis, or postmenopausal women), and patients with IMNM who are known to have difficult-to-treat myositis.
- When treatment is initiated with prednisone alone, a second-line agent is added in patients who fail to significantly improve after 2–4 months of treatment or in those who cannot be tapered to a low dose of prednisone.
- Many patients with IMNM do not respond to prednisone alone or even prednisone plus a second-line agent in combination.
- Many require triple therapy with prednisone, methotrexate, and IVIG and, if this fails, rituximab.
- In our experience and that of others, anti-HMGCR myopathy often responds to monotherapy with IVIG.
- Unfortunately, IBM does not typically respond to any known immunotherapy.
- The mainstay of treatment is physical and occupational therapy to improve function, and swallowing therapy (and sometimes esophageal dilation or cricopharyngeal myotomy) in those with dysphagia.
10. KEY PEARLS & CLINICAL TRAPS¶
- IBM is the most common cause of myopathy in those aged >50.
- IBM is characterized by asymmetric weakness of finger/wrist flexors and quadriceps, and poor response to immunotherapy.
- IBM is not typically responsive to immunotherapy; treatment focuses on physical/occupational therapy and swallowing management.
- Malignancy screening is crucial in DM, PM, and IMNM, especially with anti-TIF1-γ or anti-NXP2 antibodies.
- ILD is associated with anti-MDA5 DM and ASyS; spiral chest CT is best for honeycomb pattern.
- Anti-HMGCR myopathy does not improve when statins are discontinued.
- Anti-SRP myopathies are notable for the presence of anti-SRP antibodies and a typically subacute, aggressive, and relatively refractory course.
- A muscle biopsy should be performed in every case of suspected PM to exclude IBM (if not clinically apparent) and other causes of myopathy.
- Patients with severe muscle pain, subjective weakness, and fatigue with normal strength and function on examination are not likely to have an IM.
- Polymyalgia rheumatica should be considered in older individuals with an elevated ESR or CRP but normal CK and EMG.
- A heliotrope rash combined with Gottron papules and dilated nailfold capillaries is diagnostic for DM.
- The presence of atrophy and weakness of the flexor forearm muscles and quadriceps in a person aged >50 years is most likely IBM.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 377-2 Perifascicular atrophy and myxovirus resistance protein A (MxA) expression in dermatomyositis. A. therapies. Perifascicular myofibers (black arrows) bordering on disrupted perimysial connective tissue are atrophic and basophilic series of “PM” on hematoxylin and eosin (H&E) stains. B. Perifascicular myofibers (white arrows) show intense staining for MxA protein along a gradient from superficial to deep; all capillaries show intense MxA expression (white arrowheads). who actually myopathies layer of keratinocytes is most damaged; the inflammatory infiltrate is phies) that do not respond to typically absent or minimal and, when present, is located mainly at the nostic features are cancer, increased age, lung border zone of the dermis and epidermis. and late or previously inadequate treatment. The pathogenesis of DM was traditionally attributed to an — Figure 377-1: Cutaneous manifestations of dermatomyositis showing macular erythema plaques (Gottron sign) and erythematous papules on extensor surfaces, V-sign on anterior neck/chest, Shawl sign on posterior neck/shoulders, and nail bed capillary changes.
Figure 2¶

Caption: FIGURE 377-1 Cutaneous manifestations of dermatomyositis. A. Macular erythema a erythematous papules (Gottron papules) on extensor surface of fingers and B. elbow. C. over anterior neck and chest (V-sign) and D. the posterior neck, shoulder, and upper changes with dilated capillaries. — Figure 377-2: Histology of dermatomyositis demonstrating perifascicular myofiber atrophy (H&E stain) and intense staining for myxovirus resistance protein A (MxA) along a gradient from superficial to deep capillaries.
Figure 3¶

Caption: FIGURE 377-6 Pathology of myositis with anti-Jo-1 antibodies (antisynthetase associated with perimysial connective tissue is edematous and fragmented in tissue intensely stains red with alkaline phosphatase stain (arrowhead). C. sarcolemma of nonnecrotic perifascicular muscle fibers (open arrow). ratio with an increased CD8 count. Needle EMG may demonstrate large-amplitude, long-duration motor unit potentials that can be mis- interpreted as neurogenic but reflect the chronicity of the myopathy. Muscle MRI may show a predilection for involvement of the flexor digitorum profundus in the arms and the vastus medialis and lateralis — Figure 377-3: Skeletal muscle MRI with short T1 inversion recovery (STIR) imaging in polymyositis showing bright signal indicative of edema/inflammation in the rectus femoris muscle, contrasting with inclusion body myositis which shows selective involvement of vastus lateralis/medialis with sparing of rectus femoris.
Figure 4¶

Caption: FIGURE 377-8 Pathology of inclusion body myositis. A. Scattered muscle fibers with eosin stain. B. Cytochrome oxidase stain demonstrates an increased number of p62 within a muscle fiber (thick arrow). D. Electromicroscopy reveals 15- to 21-nm — Figure 377-4: Pathology of polymyositis demonstrating endomysial inflammatory infiltrates surrounding nonnecrotic muscle fibers.
Figure 5¶

Caption: FIGURE 377-6 Pathology of myositis with anti-Jo-1 antibodies (antisynthetase associated with perimysial connective tissue is edematous and fragmented in tissue intensely stains red with alkaline phosphatase stain (arrowhead). C. sarcolemma of nonnecrotic perifascicular muscle fibers (open arrow). ratio with an increased CD8 count. Needle EMG may demonstrate large-amplitude, long-duration motor unit potentials that can be mis- interpreted as neurogenic but reflect the chronicity of the myopathy. Muscle MRI may show a predilection for involvement of the flexor digitorum profundus in the arms and the vastus medialis and lateralis — Figure 377-5: Pathology of immune-mediated necrotizing myopathy showing scattered necrotic fibers with inflammatory infiltrate confined to fibers undergoing myophagocytosis along with regenerating fibers.
Figure 6¶

Caption: FIGURE 377-4 Pathology of polymyositis. Muscle biopsy demonstrates endomysial infiltrates surrounding nonnecrotic muscle fibers. — Figure 377-6: Pathology of myositis with anti-Jo-1 antibodies (antisynthetase syndrome) showing perifascicular/perimysial muscle fiber atrophy, edematous fragmented connective tissue, alkaline phosphatase staining, and membrane attack complex (MAC) deposits on sarcolemma.
Figure 7¶

Caption: FIGURE 377-2 Perifascicular atrophy and myxovirus resistance protein A (MxA) expression in dermatomyositis. A. therapies. Perifascicular myofibers (black arrows) bordering on disrupted perimysial connective tissue are atrophic and basophilic series of “PM” on hematoxylin and eosin (H&E) stains. B. Perifascicular myofibers (white arrows) show intense staining for MxA protein along a gradient from superficial to deep; all capillaries show intense MxA expression (white arrowheads). who actually myopathies layer of keratinocytes is most damaged; the inflammatory infiltrate is phies) that do not respond to typically absent or minimal and, when present, is located mainly at the nostic features are cancer, increased age, lung border zone of the dermis and epidermis. and late or previously inadequate treatment. The pathogenesis of DM was traditionally attributed to an — Figure 377-7: Muscle manifestations of inclusion body myositis showing finger flexor weakness (subtle to severe), ventral forearm atrophy, medial thigh atrophy, and advanced fibrous replacement of vastus medialis.
Figure 8¶
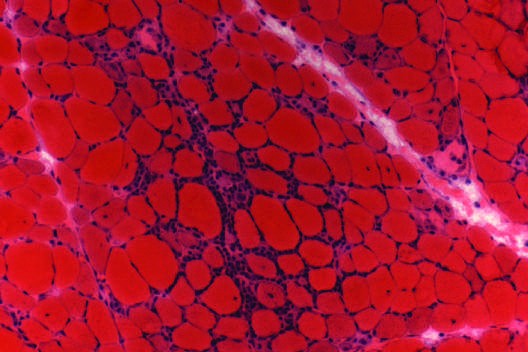
Caption: FIGURE 377-4 Pathology of polymyositis. Muscle biopsy demonstrates endomysial infiltrates surrounding nonnecrotic muscle fibers. — Figure 377-8: Pathology of inclusion body myositis showing scattered muscle fibers with rimmed vacuoles, eosinophilic inclusions, cytochrome oxidase-negative fibers, p62-positive cytoplasmic inclusions, and 15- to 21-nm tubulofilamentous inclusions within myonuclei.
Figure 9¶

Caption: Figure 377-9: Overview of inflammatory myopathies diagnostic approach and differential diagnosis (inferred from chapter context).
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.