Chapter 428 | Part 12: Endocrinology and Metabolism: The Porphyrias¶
Chapter 428 | Part 12: Endocrinology and Metabolism · Part 12 – Endocrinology & Metabolism
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- Porphyrias are metabolic disorders resulting from deficiency or increased activity of a specific enzyme in the heme biosynthetic pathway.
- Classification: Hepatic (liver origin) vs. Erythropoietic (erythrocyte origin); Acute (neurovisceral) vs. Cutaneous (photosensitivity).
- Acute hepatic porphyrias (AIP, HCP, VP) present with abdominal pain, peripheral neuropathy, and mental disturbances, often precipitated by drugs, dieting, or hormones.
- Cutaneous porphyrias (PCT, CEP, EPP, XLP) present with photosensitivity; CEP is severe and blistering, EPP is non-blistering and painful.
- Diagnosis: Spot urine PBG is sensitive and specific for acute attacks; specific porphyrin profiles (urine, plasma, feces) distinguish cutaneous types.
- Genetic testing is required for definitive diagnosis and prenatal counseling; molecular analysis identifies specific gene mutations.
- Acute attacks treated with hemin or givosiran; maintenance therapy involves avoiding precipitating factors and specific drugs.
- Pregnancy: Acute porphyrias usually well-tolerated; maintenance chelation (zinc) safe during pregnancy; avoid other chelators due to teratogenicity.
- Epidemiology: AIP ~1 in 20,000 Caucasians; EPP ~1 in 75,000 to 1 in 150,000; CEP very rare (~200 cases worldwide).
- Precipitating factors: Drugs (induce CYP enzymes), alcohol, fasting, steroids, hormonal changes (estrogens, progesterone).
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 Classification
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Regulation of Heme Biosynthesis
- 4. CLINICAL FEATURES
- 4.1 Acute Hepatic Porphyrias
- 4.2 Cutaneous Porphyrias
- 4.3 Specific Porphyria Presentations
- 5. DIFFERENTIAL DIAGNOSIS
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Diagnostic Algorithm
- 7. MANAGEMENT & TREATMENT
- 7.1 Acute Attack Management
- 7.2 Maintenance Therapy
- 8. PROGNOSIS & COMPLICATIONS
- 9. SPECIAL CONSIDERATIONS
- 10. KEY PEARLS & CLINICAL TRAPS
- Flowcharts & Algorithms
- Figures & Illustrations
📋 Figures in This Chapter¶
1. DEFINITION & OVERVIEW¶
- The porphyrias are metabolic disorders, each resulting from the deficiency or increased activity of a specific enzyme in the heme biosynthetic pathway.
- These enzyme disorders are inherited as autosomal dominant, autosomal recessive, or X-linked traits, with the exception of porphyria cutanea tarda (PCT), which is usually sporadic.
- The porphyrias are classified as either hepatic or erythropoietic, depending on the primary site of overproduction and accumulation of their respective porphyrin precursors or porphyrins.
- Some porphyrias have overlapping features; for example, PCT is hepatic and presents with blistering cutaneous photosensitivity, which is typically characteristic of the erythropoietic porphyrias.
- The major manifestations of the acute hepatic porphyrias are neurologic, including neuropathic abdominal pain, peripheral motor neuropathy, and mental disturbances, with attacks often precipitated by dieting, certain porphyrinogenic drugs, and hormonal changes.
- While hepatic porphyrias are symptomatic primarily in adults, rare homozygous variants of the autosomal dominant hepatic porphyrias usually manifest clinically prior to puberty.
- In contrast, the erythropoietic porphyrias usually present at birth or in early childhood with cutaneous photosensitivity or, in the case of congenital erythropoietic porphyria (CEP), even in utero as nonimmune hydrops fetalis.
- Cutaneous sensitivity to sunlight results from excitation of excess porphyrins in the skin by long-wave ultraviolet light, leading to cell damage, scarring, and disfigurement.
- Thus, the porphyrias are metabolic disorders in which environmental, physiologic, and genetic factors interact to cause disease.
- Because many symptoms of the porphyrias are nonspecific, diagnosis is often delayed.
- Laboratory measurement of porphyrin precursors (5'-aminolevulinic acid [ALA] and porphobilinogen [PBG]) in the urine or porphyrins in the urine, plasma, erythrocytes, or feces is required to confirm or exclude the various types of porphyria.
- A definite diagnosis requires demonstration of the specific gene defect.
- The genes encoding all the heme biosynthetic enzymes have been characterized, permitting identification of the mutations causing each porphyria.
- Molecular genetic analyses now make it possible to provide precise heterozygote or homozygote identification and prenatal diagnoses in families with known mutations.
- In addition to recent reviews of the porphyrias, informative and up-to-date websites are sponsored by the United Porphyrias Association and the European Porphyria Network.
- An extensive list of unsafe and safe drugs for individuals with acute porphyrias is provided at the Drug Database for Acute Porphyrias.
1.1 Classification¶
- The porphyrias can be classified as either hepatic or erythropoietic, depending on whether the heme biosynthetic intermediates that accumulate arise initially from the liver or developing erythrocytes.
- Alternatively, they are classified as acute or cutaneous, based on their clinical manifestations.
- Three of the five hepatic porphyrias—AIP, HCP, and VP—usually present during adult life with acute attacks of neurologic manifestations and elevated levels of the porphyrin precursors, ALA and PBG, and are thus classified as acute hepatic porphyrias.
- Patients with ADP have presented in infancy and adolescence and typically have elevated ALA with normal or slightly elevated PBG levels.
- The fifth hepatic disorder, PCT, presents with blistering skin lesions.
- HCP and VP also may have cutaneous manifestations similar to PCT.
- The erythropoietic porphyrias—CEP, EPP, and XLP—are characterized by elevations of porphyrins in bone marrow and erythrocytes and present with cutaneous photosensitivity.
- The skin lesions in CEP resemble PCT but are usually much more severe, whereas EPP and XLP cause a more immediate, severe, painful, and nonblistering type of photosensitivity.
- EPP is the most common porphyria to cause symptoms before puberty.
- About 20% of EPP patients develop minor abnormalities of liver function, with up to ~5% developing hepatic complications that can lead to liver failure requiring liver transplantation.
- XLP has a clinical presentation similar to EPP causing photosensitivity and liver disease.
2. EPIDEMIOLOGY¶
- The porphyrias are panethnic metabolic diseases that affect individuals globally.
- The acute hepatic porphyrias—acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP)—are autosomal dominant disorders.
- The frequency of AIP, the most common acute hepatic porphyria, is ~1 in 20,000 among Caucasian individuals of Western European ancestry.
- It is particularly frequent in Scandinavians, where the frequency in Sweden is ~1 in 10,000.
- VP is known as particularly frequent in South Africa, and its high prevalence (>10,000 affected patients) is in part due to a genetic 'founder effect.'
- The autosomal recessive acute hepatic porphyria, ALA-dehydratase-deficient porphyria (ADP), is extremely rare, and <20 patients have been identified worldwide.
- The erythropoietic protoporphyrias—CEP, erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLP)—also are panethnic.
- EPP is likely the most common porphyria, while CEP is very rare with about 200 reported cases worldwide.
- The frequency of EPP varies globally since most patients have the common low expression allele, which ranges in frequency in different populations.
- This allele rarely occurs in Africans, is present in ~10% of the Caucasians, and is frequent (~30%) in the Japanese.
- The second enzyme, ALA-dehydratase, catalyzes the condensation of two molecules of ALA to form PBG.
- Hydroxymethylbilane synthase (HMB-synthase; also known as PBG-deaminase) catalyzes the head-to-tail condensation of four PBG molecules by a series of housekeeping deaminations to form the linear tetrapyrrole, HMB.
- Uroporphyrinogen III synthase (URO-synthase) catalyzes the rearrangement and rapid cyclization of HMB to form the asymmetric, physiologic, octacarboxylate porphyrinogen, uroporphyrinogen (URO'gen) III.
- The fifth enzyme in the pathway, uroporphyrinogen decarboxylase (URO-decarboxylase), catalyzes the sequential removal of the four carboxyl groups from the acetic acid side chains of URO'gen III to form coproporphyrinogen (COPRO'gen) III, a tetracarboxylate porphyrinogen.
- This compound then enters the mitochondrion via a specific transporter, where COPRO-oxidase, the sixth enzyme, catalyzes the decarboxylation of two of the four propionic acid groups to form the two vinyl groups of protoporphyrinogen (PROTO'gen) IX, a decarboxylate porphyrinogen.
- Next, PROTO-oxidase oxidizes PROTO'gen to protoporphyrin IX by the removal of six hydrogen atoms.
- The product of the reaction is a porphyrin (oxidized form), in contrast to the preceding tetrapyrrole intermediates, which are porphyrinogens (reduced forms).
- Finally, ferrous iron is inserted into protoporphyrin IX to form heme, a reaction catalyzed by the eighth enzyme in the pathway, FECH (also known as heme synthase or protoheme ferrolyase).
- PCT, which is typically sporadic, occurs more frequently in countries in which its predisposing risk factors such as hepatitis C and HIV are more prevalent.
- The reported prevalence of EPP in the Caucasian population ranges from 1 in ~75,000 to 1 in ~150,000.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- Heme biosynthesis involves eight enzymatic steps in the conversion of glycine and succinyl-CoA to heme.
- These eight enzymes are encoded by nine genes, as the first enzyme in the pathway, ALA-synthase, has two genes that encode unique housekeeping (ALAS1) and erythroid-specific (ALAS2) isozymes.
- The first and last three enzymes in the pathway are located in the mitochondria, whereas the other four are in the cytosol.
- Heme is required for a variety of hemoproteins such as hemoglobin, myoglobin, respiratory cytochromes, and the cytochrome P450 (CYP) enzymes.
- Hemoglobin synthesis in erythroid precursor cells accounts for ~85% of daily heme synthesis in humans.
- Hepatocytes account for most of the rest, primarily for the synthesis of CYPs, which are especially abundant in the liver endoplasmic reticulum, and turn over more rapidly than many other hemoproteins, such as the mitochondrial respiratory cytochromes.
- As shown in the pathway, the pathway intermediates are the porphyrin precursors, ALA and PBG, and porphyrins (mostly in their reduced forms, porphyrinogens).
- At least in humans, these intermediates do not accumulate in significant amounts under normal conditions or have important physiologic functions.
- The first enzyme, ALA-synthase, catalyzes the condensation of glycine, activated by pyridoxal phosphate and succinyl-coenzyme A, to form ALA.
- In the liver, this rate-limiting enzyme can be induced by a variety of drugs, steroids, and other chemicals.
- Distinct nonerythroid (e.g., housekeeping) and erythroid-specific forms of ALA-synthase are encoded by separate genes located on chromosome 3p21.1 (ALAS1) and Xp11.2 (ALAS2), respectively.
- Defects in the erythroid gene ALAS2 that decrease its activity cause an X-linked sideroblastic anemia (XLSA).
- Gain-of-function mutations in the last exon (11) of ALAS2 that increase its activity cause an X-linked form of EPP, known as XLP.
- The second enzyme, ALA-dehydratase, catalyzes the condensation of two molecules of ALA to form PBG.
- Hydroxymethylbilane synthase (HMB-synthase; also known as PBG-deaminase) catalyzes the head-to-tail condensation of four PBG molecules by a series of housekeeping deaminations to form the linear tetrapyrrole, HMB.
- Uroporphyrinogen III synthase (URO-synthase) catalyzes the rearrangement and rapid cyclization of HMB to form the asymmetric, physiologic, octacarboxylate porphyrinogen, uroporphyrinogen (URO'gen) III.
- The fifth enzyme in the pathway, uroporphyrinogen decarboxylase (URO-decarboxylase), catalyzes the sequential removal of the four carboxyl groups from the acetic acid side chains of URO'gen III to form coproporphyrinogen (COPRO'gen) III, a tetracarboxylate porphyrinogen.
- This compound then enters the mitochondrion via a specific transporter, where COPRO-oxidase, the sixth enzyme, catalyzes the decarboxylation of two of the four propionic acid groups to form the two vinyl groups of protoporphyrinogen (PROTO'gen) IX, a decarboxylate porphyrinogen.
- Next, PROTO-oxidase oxidizes PROTO'gen to protoporphyrin IX by the removal of six hydrogen atoms.
- The product of the reaction is a porphyrin (oxidized form), in contrast to the preceding tetrapyrrole intermediates, which are porphyrinogens (reduced forms).
- Finally, ferrous iron is inserted into protoporphyrin IX to form heme, a reaction catalyzed by the eighth enzyme in the pathway, FECH (also known as heme synthase or protoheme ferrolyase).
- Regulation of heme synthesis differs in the two major heme-forming tissues, the liver and erythron.
- In the liver, the concentration of 'free' heme regulates the synthesis and mitochondrial translocation of the housekeeping form of ALA-synthase 1.
- Heme represses the synthesis of the ALA-synthase 1 messenger RNA (mRNA) and interferes with the transport of the enzyme from the cytosol into mitochondria.
- Hepatic ALA-synthase 1 is increased by many of the same chemicals that induce the CYP enzymes in the endoplasmic reticulum of the liver.
- Because most of the heme in the liver is used for the synthesis of CYP enzymes, hepatic ALA-synthase 1 and the CYPs are regulated in a coordinated fashion, and many drugs that induce hepatic ALA-synthase 1 also induce CYP gene expression.
- The other hepatic heme biosynthetic enzymes are presumably expressed at constant levels, although their relative activities and kinetic properties differ.
- For example, normal individuals have high activities of ALA-dehydratase but low activities of HMB-synthase, the latter being the second rate-limiting step in the pathway.
- In the erythron, novel regulatory mechanisms allow for the production of the very large amounts of heme needed for hemoglobin synthesis.
- The response to stimuli for hemoglobin synthesis occurs during cell differentiation, leading to an increase in cell number.
- In contrast, the erythroid-specific ALA-synthase 2 is expressed at higher levels than the housekeeping enzyme, and erythroid-specific control mechanisms regulate other pathway enzymes as well as iron transport into erythroid cells.
- Separate erythroid-specific and nonerythroid or 'housekeeping' transcripts are known for the first four enzymes in the pathway.
- As noted above, housekeeping- and erythroid-specific ALA-synthases are encoded by genes on different chromosomes, but for each of the next three genes in the pathway, both erythroid and nonerythroid transcripts are transcribed by alternative promoters from their single respective genes.
3.1 Regulation of Heme Biosynthesis¶
- In the liver, the concentration of 'free' heme regulates the synthesis and mitochondrial translocation of the housekeeping form of ALA-synthase 1.
- Heme represses the synthesis of the ALA-synthase 1 messenger RNA (mRNA) and interferes with the transport of the enzyme from the cytosol into mitochondria.
- Hepatic ALA-synthase 1 is increased by many of the same chemicals that induce the CYP enzymes in the endoplasmic reticulum of the liver.
- Because most of the heme in the liver is used for the synthesis of CYP enzymes, hepatic ALA-synthase 1 and the CYPs are regulated in a coordinated fashion, and many drugs that induce hepatic ALA-synthase 1 also induce CYP gene expression.
- The other hepatic heme biosynthetic enzymes are presumably expressed at constant levels, although their relative activities and kinetic properties differ.
- For example, normal individuals have high activities of ALA-dehydratase but low activities of HMB-synthase, the latter being the second rate-limiting step in the pathway.
- In the erythron, novel regulatory mechanisms allow for the production of the very large amounts of heme needed for hemoglobin synthesis.
- The response to stimuli for hemoglobin synthesis occurs during cell differentiation, leading to an increase in cell number.
- In contrast, the erythroid-specific ALA-synthase 2 is expressed at higher levels than the housekeeping enzyme, and erythroid-specific control mechanisms regulate other pathway enzymes as well as iron transport into erythroid cells.
- Separate erythroid-specific and nonerythroid or 'housekeeping' transcripts are known for the first four enzymes in the pathway.
- As noted above, housekeeping- and erythroid-specific ALA-synthases are encoded by genes on different chromosomes, but for each of the next three genes in the pathway, both erythroid and nonerythroid transcripts are transcribed by alternative promoters from their single respective genes.
4. CLINICAL FEATURES¶
- Acute Hepatic Porphyrias: An acute hepatic porphyria should be suspected in patients with neurovisceral symptoms after puberty.
- Symptoms include acute abdominal pain, nausea, vomiting, tachycardia, hypertension, and motor neuropathy.
- As these symptoms are common, other causes should be ruled out.
- The diagnosis is made by measuring urinary porphyrin precursors (ALA and PBG) in a spot sample of urine.
- Urinary PBG is always increased during acute attacks of AIP, HCP, and VP and is not substantially increased in any other medical condition.
- Therefore, this measurement is both sensitive and specific.
- Results from spot (single-void) urine specimens are highly informative because very substantial increases in PBG are expected during acute attacks of porphyria.
- A 24-h collection is unnecessary.
- The same spot urine specimen should be saved for quantitative determination of ALA, PBG, and creatinine, in order to confirm the qualitative PBG result and also to detect patients with ADP.
- Urinary porphyrins may remain increased longer than porphyrin precursors in HCP and VP.
- Therefore, it is useful to measure total urinary porphyrins in the same sample, keeping in mind that urinary porphyrin increases are often nonspecific.
- Measurement of urinary porphyrins alone should be avoided for screening, because these may be increased in disorders other than porphyrias, such as chronic liver disease, and misdiagnoses of porphyria can result from minimal increases in urinary porphyrins that have no diagnostic significance.
- Measurement of erythrocyte HMB-synthase is not useful as a first-line test.
- Moreover, the enzyme activity is not decreased in all AIP patients, a borderline low normal value is not diagnostic, and the enzyme is not deficient in other acute porphyrias.
- More extensive testing is justified when an initial test is positive.
- A substantial increase in PBG may be due to AIP, HCP, or VP.
- These acute porphyrias can be distinguished by measuring urinary porphyrins (using the same spot urine sample), fecal porphyrins, and plasma porphyrins.
- Assays for COPRO-oxidase or PROTO-oxidase are not available for clinical testing.
- More specifically, mutation analysis by sequencing the genes encoding HMB-synthase, COPRO-oxidase, and PROTO-oxidase will detect almost all disease-causing mutations and is diagnostic even when the levels of urinary ALA and PBG have returned to normal or near normal.
- Cutaneous Porphyrias: Blistering skin lesions due to porphyria are virtually always accompanied by increases in total plasma porphyrins.
- A fluorometric method is preferred, because the plasma porphyrins in VP are mostly covalently linked to plasma proteins and may be less readily detected by high-performance liquid chromatography (HPLC).
- The normal range for plasma porphyrins is somewhat increased in patients with end-stage renal disease.
- Although a total plasma porphyrin determination will usually detect EPP and XLP, an erythrocyte protoporphyrin determination is more sensitive.
- Increases in erythrocyte protoporphyrin occur in many other conditions.
- Therefore, the diagnosis of EPP must be confirmed by showing a predominant increase in free protoporphyrin rather than zinc protoporphyrin.
- In XLP, both free and zinc protoporphyrin are markedly increased.
- Interpretation of laboratory reports can be difficult, because the term free erythrocyte protoporphyrin sometimes actually represents zinc protoporphyrin.
- The various porphyrias that cause blistering skin lesions can be differentiated by measuring porphyrins in urine, feces, and plasma.
- The porphyrias should be confirmed by genetic testing and the demonstration of the causative pathogenic variant.
- It is often difficult to diagnose or 'rule out' porphyria in patients who have had suggestive symptoms months or years in the past and in relatives of patients with acute porphyrias, because porphyrin precursors and porphyrins may be normal.
- In those situations, detection of the specific gene mutation in the index case can make the diagnosis and facilitate the diagnosis and genetic counseling of at-risk relatives.
- With the increased access and accuracy of genetic testing, this often precedes secondary biochemical testing in clinical practice.
- Consultation with a specialist laboratory and physician will assist in selecting the heme biosynthetic gene or genes to be sequenced.
- Clinical Features of ADP: The clinical presentation depends on the amount of residual ALA-dehydratase activity.
- Four of the documented patients were male adolescents with symptoms resembling those of AIP, including abdominal pain and neuropathy.
- One patient was an infant with more severe disease, including failure to thrive beginning at birth.
- The earlier age of onset and more severe manifestations in this patient reflect a more significant deficiency of ALA-dehydratase activity.
- Another patient developed an acute motor polyneuropathy at age 63 that was associated with a myeloproliferative disorder.
- He was heterozygous for an δ-aminolevulinic acid dehydratase (ALAD) mutation that presumably was present in erythroblasts that underwent clonal expansion due to the bone marrow malignancy.
- Clinical Features of AIP: Induction and increased expression of the rate-limiting hepatic gene ALAS1 in heterozygotes who have half-normal HMB-synthase activity is thought to underlie the acute attacks in AIP.
- The disorder remains latent (or asymptomatic) in the great majority of those who are heterozygous for pathogenic HMBS mutations, and this is almost always the case prior to puberty.
- In patients with no history of acute symptoms, porphyrin precursor excretion is usually normal, suggesting that half-normal hepatic HMB-synthase activity is sufficient and that hepatic ALA-synthase activity is not increased.
- However, under conditions where heme synthesis is increased in the liver, half-normal HMB-synthase activity may become limiting, and ALA, PBG, and other heme pathway intermediates may accumulate and be excreted in the urine.
- Common precipitating factors include endogenous and exogenous steroids, porphyrinogenic drugs, alcohol ingestion, and low-calorie diets, usually instituted for weight loss.
- The fact that AIP is almost always latent before puberty suggests that adult levels of steroid hormones are important for clinical expression.
- Symptoms are more common in women, suggesting a role for estrogens or progestins.
- Premenstrual attacks are probably due to increasing endogenous progesterone during the luteal phase of the menstrual cycle.
- Acute porphyrias are sometimes exacerbated by exogenous steroids, including oral contraceptive preparations containing progestins.
- Surprisingly, pregnancy is usually well tolerated, suggesting that beneficial metabolic changes may ameliorate the effects of high levels of progesterone.
- Extensive lists of unsafe and safe drugs are available on websites sponsored by the United Porphyria Association and the European Porphyria Network, and at the Drug Database for Acute Porphyrias website.
- Reduced intake of calories and carbohydrate, as may occur with illness or attempts to lose weight, can also increase porphyrin precursor excretion and induce attacks of porphyria.
- Clinical Features of Homozygous AIP: Homozygous dominant AIP (HD-AIP) is a rare form of AIP in which patients inherit HMBS mutations from each of their heterozygous parents and, therefore, have very low (<2%) enzyme activity.
- The disease has been described in a Dutch girl, two young British siblings, and a Spanish boy.
- In these homozygous affected patients, the disease presented in infancy with failure to thrive, developmental delay, bilateral cataracts, and/or hepatosplenomegaly.
- Urinary ALA and PBG concentrations were markedly elevated.
- All of these patients' HMBS mutations (R167W, R167Q, and R172Q) were in exon 10 within five bases of each other.
- Studies of the brain magnetic resonance images (MRIs) of children with homozygous AIP have suggested damage primarily in white matter that was myelinated postnatally, while tracks that myelinated prenatally were normal.
- Most children with homozygous AIP die at an early age.
- Recently, later-onset HD-AIP was described in an adult with leukoencephalopathy.
- Clinical Features of HCP and VP: These acute porphyrias can be distinguished by measuring urinary porphyrins (using the same spot urine sample), fecal porphyrins, and plasma porphyrins.
- Assays for COPRO-oxidase or PROTO-oxidase are not available for clinical testing.
- More specifically, mutation analysis by sequencing the genes encoding HMB-synthase, COPRO-oxidase, and PROTO-oxidase will detect almost all disease-causing mutations and is diagnostic even when the levels of urinary ALA and PBG have returned to normal or near normal.
- Clinical Features of PCT: PCT, which is typically sporadic, occurs more frequently in countries in which its predisposing risk factors such as hepatitis C and HIV are more prevalent.
- The reported prevalence of EPP in the Caucasian population ranges from 1 in ~75,000 to 1 in ~150,000.
- Clinical Features of CEP: The skin lesions in CEP resemble PCT but are usually much more severe.
- Clinical Features of EPP: EPP is the most common porphyria to cause symptoms before puberty.
- About 20% of EPP patients develop minor abnormalities of liver function, with up to ~5% developing hepatic complications that can lead to liver failure requiring liver transplantation.
- Clinical Features of XLP: XLP has a clinical presentation similar to EPP causing photosensitivity and liver disease.
4.1 Acute Hepatic Porphyrias¶
- An acute hepatic porphyria should be suspected in patients with neurovisceral symptoms after puberty.
- Symptoms include acute abdominal pain, nausea, vomiting, tachycardia, hypertension, and motor neuropathy.
- As these symptoms are common, other causes should be ruled out.
- The diagnosis is made by measuring urinary porphyrin precursors (ALA and PBG) in a spot sample of urine.
- Urinary PBG is always increased during acute attacks of AIP, HCP, and VP and is not substantially increased in any other medical condition.
- Therefore, this measurement is both sensitive and specific.
- Results from spot (single-void) urine specimens are highly informative because very substantial increases in PBG are expected during acute attacks of porphyria.
- A 24-h collection is unnecessary.
- The same spot urine specimen should be saved for quantitative determination of ALA, PBG, and creatinine, in order to confirm the qualitative PBG result and also to detect patients with ADP.
- Urinary porphyrins may remain increased longer than porphyrin precursors in HCP and VP.
- Therefore, it is useful to measure total urinary porphyrins in the same sample, keeping in mind that urinary porphyrin increases are often nonspecific.
- Measurement of urinary porphyrins alone should be avoided for screening, because these may be increased in disorders other than porphyrias, such as chronic liver disease, and misdiagnoses of porphyria can result from minimal increases in urinary porphyrins that have no diagnostic significance.
- Measurement of erythrocyte HMB-synthase is not useful as a first-line test.
- Moreover, the enzyme activity is not decreased in all AIP patients, a borderline low normal value is not diagnostic, and the enzyme is not deficient in other acute porphyrias.
- More extensive testing is justified when an initial test is positive.
- A substantial increase in PBG may be due to AIP, HCP, or VP.
- These acute porphyrias can be distinguished by measuring urinary porphyrins (using the same spot urine sample), fecal porphyrins, and plasma porphyrins.
- Assays for COPRO-oxidase or PROTO-oxidase are not available for clinical testing.
- More specifically, mutation analysis by sequencing the genes encoding HMB-synthase, COPRO-oxidase, and PROTO-oxidase will detect almost all disease-causing mutations and is diagnostic even when the levels of urinary ALA and PBG have returned to normal or near normal.
4.2 Cutaneous Porphyrias¶
- Blistering skin lesions due to porphyria are virtually always accompanied by increases in total plasma porphyrins.
- A fluorometric method is preferred, because the plasma porphyrins in VP are mostly covalently linked to plasma proteins and may be less readily detected by high-performance liquid chromatography (HPLC).
- The normal range for plasma porphyrins is somewhat increased in patients with end-stage renal disease.
- Although a total plasma porphyrin determination will usually detect EPP and XLP, an erythrocyte protoporphyrin determination is more sensitive.
- Increases in erythrocyte protoporphyrin occur in many other conditions.
- Therefore, the diagnosis of EPP must be confirmed by showing a predominant increase in free protoporphyrin rather than zinc protoporphyrin.
- In XLP, both free and zinc protoporphyrin are markedly increased.
- Interpretation of laboratory reports can be difficult, because the term free erythrocyte protoporphyrin sometimes actually represents zinc protoporphyrin.
- The various porphyrias that cause blistering skin lesions can be differentiated by measuring porphyrins in urine, feces, and plasma.
- The porphyrias should be confirmed by genetic testing and the demonstration of the causative pathogenic variant.
- It is often difficult to diagnose or 'rule out' porphyria in patients who have had suggestive symptoms months or years in the past and in relatives of patients with acute porphyrias, because porphyrin precursors and porphyrins may be normal.
- In those situations, detection of the specific gene mutation in the index case can make the diagnosis and facilitate the diagnosis and genetic counseling of at-risk relatives.
- With the increased access and accuracy of genetic testing, this often precedes secondary biochemical testing in clinical practice.
- Consultation with a specialist laboratory and physician will assist in selecting the heme biosynthetic gene or genes to be sequenced.
4.3 Specific Porphyria Presentations¶
- ADP: The clinical presentation depends on the amount of residual ALA-dehydratase activity.
- Four of the documented patients were male adolescents with symptoms resembling those of AIP, including abdominal pain and neuropathy.
- One patient was an infant with more severe disease, including failure to thrive beginning at birth.
- The earlier age of onset and more severe manifestations in this patient reflect a more significant deficiency of ALA-dehydratase activity.
- Another patient developed an acute motor polyneuropathy at age 63 that was associated with a myeloproliferative disorder.
- He was heterozygous for an δ-aminolevulinic acid dehydratase (ALAD) mutation that presumably was present in erythroblasts that underwent clonal expansion due to the bone marrow malignancy.
- AIP: Induction and increased expression of the rate-limiting hepatic gene ALAS1 in heterozygotes who have half-normal HMB-synthase activity is thought to underlie the acute attacks in AIP.
- The disorder remains latent (or asymptomatic) in the great majority of those who are heterozygous for pathogenic HMBS mutations, and this is almost always the case prior to puberty.
- In patients with no history of acute symptoms, porphyrin precursor excretion is usually normal, suggesting that half-normal hepatic HMB-synthase activity is sufficient and that hepatic ALA-synthase activity is not increased.
- However, under conditions where heme synthesis is increased in the liver, half-normal HMB-synthase activity may become limiting, and ALA, PBG, and other heme pathway intermediates may accumulate and be excreted in the urine.
- Common precipitating factors include endogenous and exogenous steroids, porphyrinogenic drugs, alcohol ingestion, and low-calorie diets, usually instituted for weight loss.
- The fact that AIP is almost always latent before puberty suggests that adult levels of steroid hormones are important for clinical expression.
- Symptoms are more common in women, suggesting a role for estrogens or progestins.
- Premenstrual attacks are probably due to increasing endogenous progesterone during the luteal phase of the menstrual cycle.
- Acute porphyrias are sometimes exacerbated by exogenous steroids, including oral contraceptive preparations containing progestins.
- Surprisingly, pregnancy is usually well tolerated, suggesting that beneficial metabolic changes may ameliorate the effects of high levels of progesterone.
- Extensive lists of unsafe and safe drugs are available on websites sponsored by the United Porphyria Association and the European Porphyria Network, and at the Drug Database for Acute Porphyrias website.
- Reduced intake of calories and carbohydrate, as may occur with illness or attempts to lose weight, can also increase porphyrin precursor excretion and induce attacks of porphyria.
- Homozygous AIP: Homozygous dominant AIP (HD-AIP) is a rare form of AIP in which patients inherit HMBS mutations from each of their heterozygous parents and, therefore, have very low (<2%) enzyme activity.
- The disease has been described in a Dutch girl, two young British siblings, and a Spanish boy.
- In these homozygous affected patients, the disease presented in infancy with failure to thrive, developmental delay, bilateral cataracts, and/or hepatosplenomegaly.
- Urinary ALA and PBG concentrations were markedly elevated.
- All of these patients' HMBS mutations (R167W, R167Q, and R172Q) were in exon 10 within five bases of each other.
- Studies of the brain magnetic resonance images (MRIs) of children with homozygous AIP have suggested damage primarily in white matter that was myelinated postnatally, while tracks that myelinated prenatally were normal.
- Most children with homozygous AIP die at an early age.
- Recently, later-onset HD-AIP was described in an adult with leukoencephalopathy.
- PCT: PCT, which is typically sporadic, occurs more frequently in countries in which its predisposing risk factors such as hepatitis C and HIV are more prevalent.
- CEP: The skin lesions in CEP resemble PCT but are usually much more severe.
- EPP: EPP is the most common porphyria to cause symptoms before puberty.
- About 20% of EPP patients develop minor abnormalities of liver function, with up to ~5% developing hepatic complications that can lead to liver failure requiring liver transplantation.
- XLP: XLP has a clinical presentation similar to EPP causing photosensitivity and liver disease.
5. DIFFERENTIAL DIAGNOSIS¶
- Hereditary tyrosinemia type 1 (fumarylacetoacetase deficiency) and lead intoxication should be considered in the differential diagnosis because either succinylacetone (which accumulates in hereditary tyrosinemia and is structurally similar to ALA) or lead can inhibit ALA-dehydratase, increase urinary excretion of ALA and COPRO III, and cause manifestations that resemble those of the acute porphyrias.
- Heterozygotes are clinically asymptomatic and do not excrete increased levels of ALA but can be detected by demonstration of intermediate levels of erythrocyte ALA-dehydratase activity or a specific mutation in the ALAD gene.
- To date, molecular studies of ADP patients have identified 12 pathogenic mutations, including missense mutations, splice-site mutations, and a two-base deletion in the ALAD gene (Human Gene Mutation Database; www.hgmd.org).
- The parents in each case were not consanguineous, and the index cases had inherited a different ALAD mutation from each parent.
- Prenatal diagnosis of this disorder is possible by determination of ALA-dehydratase activity and/or gene mutations in cultured chorionic villi or amniocytes.
6. INVESTIGATIONS & DIAGNOSIS¶
- A few specific and sensitive first-line laboratory tests should be used whenever symptoms or signs suggest the diagnosis of porphyria.
- If a first-line test is significantly abnormal, more comprehensive testing should follow to establish the type of porphyria, including the specific causative gene mutation.
- Markedly elevated plasma and urinary concentrations of the porphyrin precursors, ALA and/or PBG, which originate from the liver, are especially evident during attacks of neurologic manifestations of the four acute porphyrias—ADP, AIP, HCP, and VP.
- In PCT, excess porphyrins also accumulate initially in the liver and cause chronic blistering of sun-exposed areas of the skin.
- Diagnosis is made by measuring urinary porphyrin precursors (ALA and PBG) in a spot sample of urine.
- Urinary PBG is always increased during acute attacks of AIP, HCP, and VP and is not substantially increased in any other medical condition.
- Therefore, this measurement is both sensitive and specific.
- Results from spot (single-void) urine specimens are highly informative because very substantial increases in PBG are expected during acute attacks of porphyria.
- A 24-h collection is unnecessary.
- The same spot urine specimen should be saved for quantitative determination of ALA, PBG, and creatinine, in order to confirm the qualitative PBG result and also to detect patients with ADP.
- Urinary porphyrins may remain increased longer than porphyrin precursors in HCP and VP.
- Therefore, it is useful to measure total urinary porphyrins in the same sample, keeping in mind that urinary porphyrin increases are often nonspecific.
- Measurement of urinary porphyrins alone should be avoided for screening, because these may be increased in disorders other than porphyrias, such as chronic liver disease, and misdiagnoses of porphyria can result from minimal increases in urinary porphyrins that have no diagnostic significance.
- Measurement of erythrocyte HMB-synthase is not useful as a first-line test.
- Moreover, the enzyme activity is not decreased in all AIP patients, a borderline low normal value is not diagnostic, and the enzyme is not deficient in other acute porphyrias.
- More extensive testing is justified when an initial test is positive.
- A substantial increase in PBG may be due to AIP, HCP, or VP.
- These acute porphyrias can be distinguished by measuring urinary porphyrins (using the same spot urine sample), fecal porphyrins, and plasma porphyrins.
- Assays for COPRO-oxidase or PROTO-oxidase are not available for clinical testing.
- More specifically, mutation analysis by sequencing the genes encoding HMB-synthase, COPRO-oxidase, and PROTO-oxidase will detect almost all disease-causing mutations and is diagnostic even when the levels of urinary ALA and PBG have returned to normal or near normal.
- Blistering skin lesions due to porphyria are virtually always accompanied by increases in total plasma porphyrins.
- A fluorometric method is preferred, because the plasma porphyrins in VP are mostly covalently linked to plasma proteins and may be less readily detected by high-performance liquid chromatography (HPLC).
- The normal range for plasma porphyrins is somewhat increased in patients with end-stage renal disease.
- Although a total plasma porphyrin determination will usually detect EPP and XLP, an erythrocyte protoporphyrin determination is more sensitive.
- Increases in erythrocyte protoporphyrin occur in many other conditions.
- Therefore, the diagnosis of EPP must be confirmed by showing a predominant increase in free protoporphyrin rather than zinc protoporphyrin.
- In XLP, both free and zinc protoporphyrin are markedly increased.
- Interpretation of laboratory reports can be difficult, because the term free erythrocyte protoporphyrin sometimes actually represents zinc protoporphyrin.
- The various porphyrias that cause blistering skin lesions can be differentiated by measuring porphyrins in urine, feces, and plasma.
- The porphyrias should be confirmed by genetic testing and the demonstration of the causative pathogenic variant.
- It is often difficult to diagnose or 'rule out' porphyria in patients who have had suggestive symptoms months or years in the past and in relatives of patients with acute porphyrias, because porphyrin precursors and porphyrins may be normal.
- In those situations, detection of the specific gene mutation in the index case can make the diagnosis and facilitate the diagnosis and genetic counseling of at-risk relatives.
- With the increased access and accuracy of genetic testing, this often precedes secondary biochemical testing in clinical practice.
- Consultation with a specialist laboratory and physician will assist in selecting the heme biosynthetic gene or genes to be sequenced.
6.1 Diagnostic Algorithm¶
- Step 1: Suspect acute hepatic porphyria in patients with neurovisceral symptoms after puberty (abdominal pain, nausea, vomiting, tachycardia, hypertension, motor neuropathy).
- Step 2: Measure spot urine PBG and ALA.
- Step 3: If PBG is substantially increased, consider AIP, HCP, or VP.
- Step 4: Measure urinary porphyrins, fecal porphyrins, and plasma porphyrins to distinguish between AIP, HCP, and VP.
- Step 5: If blistering skin lesions are present, measure plasma porphyrins and erythrocyte protoporphyrin.
- Step 6: Confirm diagnosis with genetic testing (mutation analysis).
- Step 7: For ADP, measure erythrocyte ALA-dehydratase activity and/or gene mutations.
- Step 8: For EPP, confirm by showing a predominant increase in free protoporphyrin rather than zinc protoporphyrin.
7. MANAGEMENT & TREATMENT¶
- Treatment of ADP acute attacks is similar to that of AIP.
- The severely affected infant referred to above was supported by hyperalimentation and periodic blood transfusions but did not respond to intravenous hemin and died after liver transplantation.
- Acute attacks: Treatment involves avoiding precipitating factors and administering hemin or givosiran.
- Maintenance: Treatment involves avoiding precipitating factors and specific drugs.
- Pregnancy: Maintenance chelation (zinc) is safe during pregnancy; avoid other chelators due to teratogenicity.
- EPP: Avoidance of sunlight is key; liver transplantation may be required for severe cases.
7.1 Acute Attack Management¶
- Treatment of seizures is difficult because most antiseizure drugs can exacerbate AIP (clonazepam may be safer than phenytoin or barbiturates).
- Hyponatremia results from hypothalamic involvement and inappropriate vasopressin secretion or from electrolyte depletion due to vomiting, diarrhea, poor intake, or excess renal sodium loss.
- When an attack resolves, abdominal pain may disappear within hours, and paresis begins to improve within days and may continue to improve over several years.
- Hemin: Administered for acute attacks.
- Givosiran: Administered subcutaneously; levels of urinary ALA and PBG decrease over a few days after administration or in a day after subcutaneous givosiran.
- Avoidance of precipitating factors: Drugs, dieting, alcohol, fasting, steroids, hormonal changes.
7.2 Maintenance Therapy¶
- Avoidance of precipitating factors: Drugs, dieting, alcohol, fasting, steroids, hormonal changes.
- Specific drugs: Use safe drugs listed on websites sponsored by the United Porphyria Association and the European Porphyria Network, and at the Drug Database for Acute Porphyrias website.
- Pregnancy: Maintenance chelation (zinc) is safe during pregnancy; avoid other chelators due to teratogenicity.
- EPP: Avoidance of sunlight is key; liver transplantation may be required for severe cases.
8. PROGNOSIS & COMPLICATIONS¶
- About 20% of EPP patients develop minor abnormalities of liver function, with up to ~5% developing hepatic complications that can lead to liver failure requiring liver transplantation.
- XLP has a clinical presentation similar to EPP causing photosensitivity and liver disease.
- Most children with homozygous AIP die at an early age.
- Recently, later-onset HD-AIP was described in an adult with leukoencephalopathy.
- Sudden death may result from sympathetic overactivity and cardiac arrhythmia.
- Progression to respiratory and bulbar paralysis and death occurs especially when the diagnosis and treatment are delayed.
9. SPECIAL CONSIDERATIONS¶
- Pregnancy: Acute porphyrias usually well-tolerated; maintenance chelation (zinc) safe during pregnancy; avoid other chelators due to teratogenicity.
- Children: Homozygous AIP presents in infancy with failure to thrive, developmental delay, bilateral cataracts, and/or hepatosplenomegaly.
- Prenatal diagnosis: Possible by determination of ALA-dehydratase activity and/or gene mutations in cultured chorionic villi or amniocytes.
10. KEY PEARLS & CLINICAL TRAPS¶
- Spot urine PBG is always increased during acute attacks of AIP, HCP, and VP and is not substantially increased in any other medical condition.
- Measurement of urinary porphyrins alone should be avoided for screening, because these may be increased in disorders other than porphyrias, such as chronic liver disease.
- Interpretation of laboratory reports can be difficult, because the term free erythrocyte protoporphyrin sometimes actually represents zinc protoporphyrin.
- The various porphyrias that cause blistering skin lesions can be differentiated by measuring porphyrins in urine, feces, and plasma.
- The porphyrias should be confirmed by genetic testing and the demonstration of the causative pathogenic variant.
- It is often difficult to diagnose or 'rule out' porphyria in patients who have had suggestive symptoms months or years in the past and in relatives of patients with acute porphyrias, because porphyrin precursors and porphyrins may be normal.
- In those situations, detection of the specific gene mutation in the index case can make the diagnosis and facilitate the diagnosis and genetic counseling of at-risk relatives.
- With the increased access and accuracy of genetic testing, this often precedes secondary biochemical testing in clinical practice.
- Consultation with a specialist laboratory and physician will assist in selecting the heme biosynthetic gene or genes to be sequenced.
Flowcharts & Algorithms¶
Reproduced from Harrison's 22nd Edition.
Flowchart 1¶
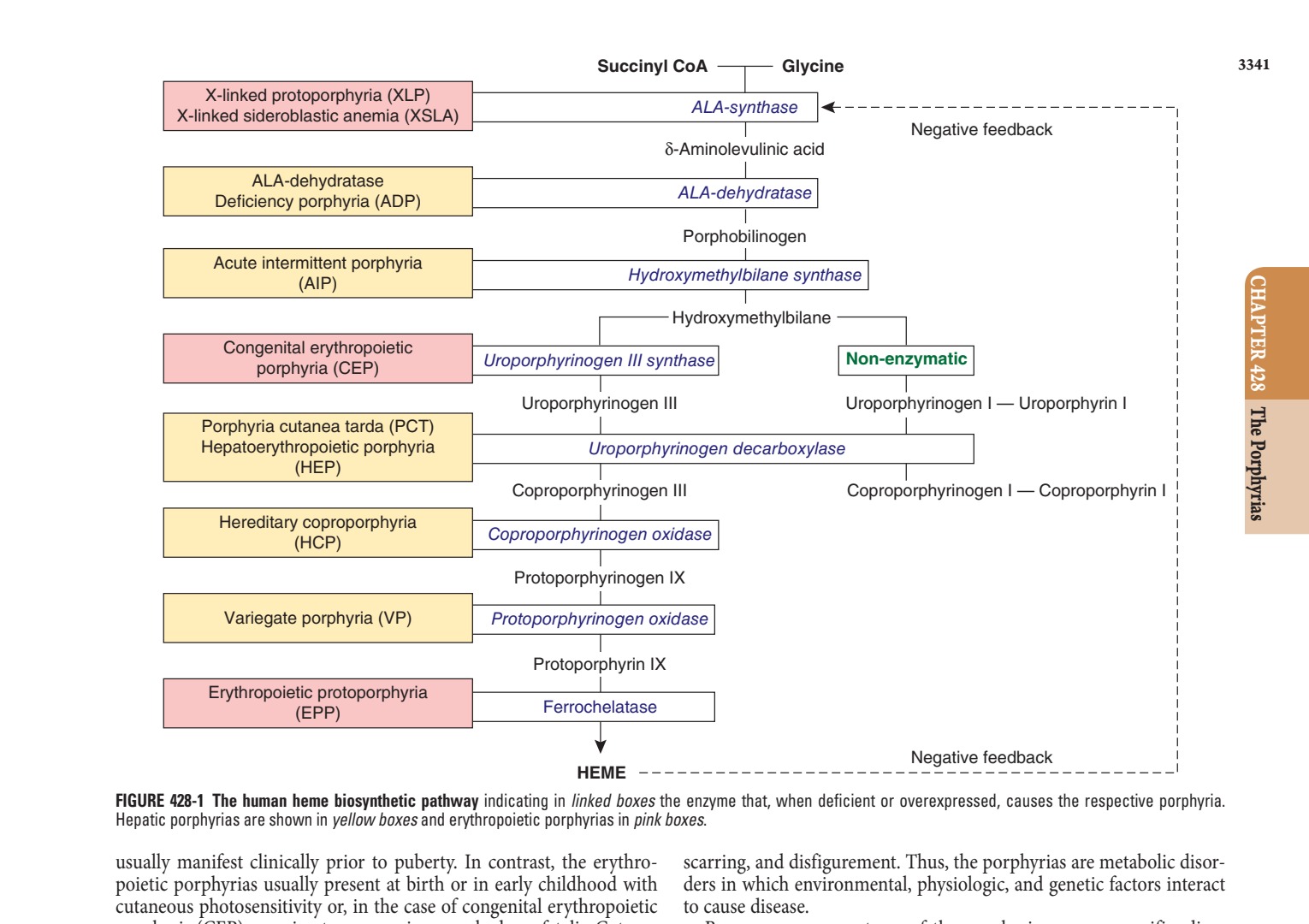
Caption: FIGURE 428-1 The human heme biosynthetic pathway indicating in linked boxes the Hepatic porphyrias are shown in yellow boxes and erythropoietic porphyrias in pink usually manifest clinically prior to puberty. In contrast, the erythro- poietic porphyrias usually present at birth or in early childhood with cutaneous photosensitivity or, in the case of congenital erythropoietic to porphyria (CEP), even in utero as nonimmune hydrops fetalis. Cutane- ous sensitivity to sunlight results from excitation of excess porphyrins in the skin by long-wave ultraviolet light, leading to cell damage,
Flowchart 2¶

Caption: FIGURE 428-2 The heme biosynthetic pathway showing the eight enzymes and their four in the cytosol.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 428-4 Erythema and edema of the hands due to acute photosensitivity in a 10-year-old boy with erythropoietic protoporphyria. (Reproduced with permission from P Poblete-Gutiérrez et al: The porphyrias: clinical presentation, diagnosis and treatment. Eur J Dermatol 16:230, 2006.) — Figure 428-1: The human heme biosynthetic pathway indicating in linked boxes the enzyme that, when deficient or overexpressed, causes the respective porphyria. Hepatic porphyrias are shown in yellow boxes and erythropoietic porphyrias in pink boxes.
Figure 2¶

Caption: FIGURE 428-3 Typical cutaneous lesions in a patient with porphyria cutanea tarda. Chronic, crusted lesions resulting from blistering due to photosensitivity on the dorsum of the hand of a patient with porphyria cutanea tarda. (Used with permission from Dr. Karl E. Anderson.) — Figure 428-2: The heme biosynthetic pathway showing the eight enzymes and their substrates and products. Four of the enzymes are localized in the mitochondria and four in the cytosol.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.