Seizures and Epilepsy¶
Chapter 436 | Part 13: Neurologic Disorders · Part 13 – Neurologic Disorders
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- A seizure is a transient occurrence of signs or symptoms due to abnormal excessive or synchronous neuronal activity in the brain.
- Epilepsy is a clinical phenomenon characterized by a risk of recurrent seizures due to a chronic, underlying process, not a single disease entity.
- The 2017 ILAE classification system categorizes seizures by onset (Focal, Generalized, Unknown) and awareness (Intact or Impaired).
- Focal seizures can evolve into generalized seizures (secondary generalization), often starting with an aura.
- Typical absence seizures show a 3-Hz spike-and-slow-wave pattern on EEG and are provoked by hyperventilation.
- Mesial temporal lobe epilepsy (MTLE) is characterized by hippocampal sclerosis and is often refractory to anticonvulsants but responsive to surgery.
- Drugs like sodium channel blockers (e.g., carbamazepine) should be avoided in SCN1A-related epilepsy (Dravet syndrome) as they can worsen seizures.
- The ketogenic diet is the gold standard treatment for refractory epilepsy due to GLUT1 deficiency.
- Febrile seizures occur in 3–5% of children, typically between 6 months and 5 years, and are not necessarily associated with epilepsy.
- Epileptogenesis refers to the process where a normal neuronal network transforms into one that is abnormally hyperexcitable.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 Classification of Seizures
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Genetic Factors and Epilepsy Syndromes
- 4. CLINICAL FEATURES
- 4.1 Focal Seizures with Intact Awareness
- 4.2 Focal Seizures with Impaired Awareness
- 4.3 Generalized Onset Seizures
- 5. DIFFERENTIAL DIAGNOSIS
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Diagnostic Criteria and Algorithms
- 7. MANAGEMENT & TREATMENT
- 7.1 Pharmacologic and Non-Pharmacologic Therapies
- 8. PROGNOSIS & COMPLICATIONS
- 9. SPECIAL CONSIDERATIONS
- 9.1 Age-Related Etiologies
- 10. KEY PEARLS & CLINICAL TRAPS
- Flowcharts & Algorithms
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🔀 Flowchart | Evaluation of the adult patient with a seizure |
| 2 | 🔀 Flowchart | Pharmacologic treatment of generalized tonic-clonic status epilepticus LEV, levetiracetam; LZP, lorazepam; MDZ,... |
| 1 | 🖼 Figure | Mesial temporal lobe epilepsy |
| 2 | 🖼 Figure | Electrographic seizures |
| 3 | 🖼 Figure | Electroencephalograms |
1. DEFINITION & OVERVIEW¶
- A seizure is defined as a transient occurrence of signs or symptoms due to abnormal excessive or synchronous neuronal activity in the brain.
- Epilepsy describes a condition in which a person has a risk of recurrent seizures due to a chronic, underlying process.
- Epilepsy refers to a clinical phenomenon rather than a single disease entity because many forms and causes exist.
- A single seizure, or recurrent seizures due to correctable or avoidable circumstances, does not necessarily have epilepsy.
- A single seizure associated with clinical or electroencephalographic features portending high risk of recurrence may establish the diagnosis of epilepsy.
- Using the definition of epilepsy as two or more unprovoked seizures, the incidence of epilepsy is ~0.3–0.5% in different populations throughout the world.
- The prevalence of epilepsy has been estimated at 5–30 persons per 1000.
1.1 Classification of Seizures¶
- The International League Against Epilepsy (ILAE) Commission on Classification and Terminology updated their approach to classification of seizures in 2017.
- This system is based on the clinical features of seizures and associated electroencephalographic findings.
- Other potentially distinctive features such as etiology or cellular substrate are not considered in this classification system.
- A fundamental principle is that seizures may be either focal or generalized.
- Focal seizures originate within networks limited to one brain region.
- Generalized seizures arise within and rapidly engage networks distributed across both cerebral hemispheres.
- Focal seizures are often associated with structural abnormalities of the brain.
- Generalized seizures may result from cellular, biochemical, or structural abnormalities with a more widespread distribution.
- There are exceptions in both cases.
- The subcategories of 'simple focal seizures' and 'complex focal seizures' have been eliminated.
- The classification emphasizes the effect on awareness (intact or impaired) and nature of the onset (motor or nonmotor).
Table 1 — TABLE 436-1 Classification of Seizures¶
| Onset | Subtypes |
|---|---|
| 1. Focal Onset | (Can be further described as having intact or impaired awareness, motor or nonmotor onset, or evolve from focal to bilateral tonic clonic) |
| 2. Generalized Onset | a. Motor: Tonic-clonic; Other motor (e.g., atonic, myoclonic) |
| 2. Generalized Onset | b. Nonmotor (absence) |
| 3. Unknown Onset | Motor, nonmotor, or unclassified |
2. EPIDEMIOLOGY¶
- The incidence of epilepsy is ~0.3–0.5% in different populations throughout the world.
- The prevalence of epilepsy has been estimated at 5–30 persons per 1000.
- ~5–10% of the population will have at least one seizure.
- The highest incidence of seizures occurs in early childhood and late adulthood.
- Febrile seizures have an overall prevalence of 3–5% and even higher in some parts of the world such as Asia.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
- Seizures are a result of a shift in the normal balance of excitation and inhibition within the CNS.
- There are differences between individuals in the susceptibility or threshold for seizures.
- Epileptogenesis is the process where the injury results in a long-lasting pathologic change in the CNS that transforms a presumably normal neuronal network into one that is abnormally hyperexcitable.
- Other processes associated with epileptogenesis include stroke, infection, neurodegeneration, and abnormalities of CNS development.
- Seizures are episodic and occur intermittently.
- Precipitants include those due to intrinsic physiologic processes (psychological or physical stress, sleep deprivation, hormonal changes) and exogenous factors (toxic substances, certain medications, intermittent photic stimulation).
3.1 Genetic Factors and Epilepsy Syndromes¶
- Epilepsy syndromes are disorders in which epilepsy is a predominant feature, and there is sufficient evidence to suggest a common underlying mechanism.
- Three important epilepsy syndromes are listed below; additional examples with a known genetic basis are shown in Table 436-2.
- Juvenile Myoclonic Epilepsy (JME) is a generalized seizure disorder of unknown cause that appears in early adolescence.
- Lennox-Gastaut syndrome occurs in children and is defined by a triad: (1) multiple seizure types (usually including generalized tonic-clonic, atonic, and atypical absence seizures); (2) an EEG with slow (<3 Hz) spike-and-wave discharges and a variety of other abnormalities; and (3) developmental delay.
- Mesial Temporal Lobe Epilepsy (MTLE) is the most common syndrome associated with focal seizures with impairment of consciousness and is an example of an epilepsy syndrome with distinctive clinical, EEG, and pathologic features.
- High-resolution magnetic resonance imaging (MRI) can detect the characteristic hippocampal sclerosis that appears to be essential in the pathophysiology of MTLE for many patients.
Table 2 — TABLE 436-2 Examples of Genes Associated with Epilepsy Syndromes¶
| Gene (Locus) | Function of Gene | Clinical Syndrome | Comments |
|---|---|---|---|
| CHRNA4 (20q13.2) | Nicotinic acetylcholine receptor subunit; mutations cause alterations in Ca2+ flux through the receptor; this may reduce the amount of GABA release in presynaptic terminals | Sleep-related hypermotor epilepsy (SHE); childhood onset; brief, nighttime seizures with prominent motor movements; often misdiagnosed as primary sleep disorder | Rare; first identified in a large Australian family; other families found to have mutations in CHRNA2 or CHRNB2, and some families appear to have mutations at other loci |
| KCNQ2 (20q13.3) | Voltage-gated potassium channel subunits; mutation in pore regions may cause a 20–40% reduction of potassium currents, which will lead to impaired repolarization | Self-limited familial neonatal epilepsy; autosomal dominant inheritance; onset in first week of life in infants who are otherwise normal; remission usually within weeks to months; long-term epilepsy in 10–15% | |
| SCN1A (2q24.3) | α-Subunit of a voltage-gated sodium channel; numerous mutations affecting sodium currents that cause either gain or loss of function; network effects appear related to expression in excitatory or inhibitory cells | Very common cause of Dravet syndrome (severe myoclonic epilepsy of infancy) and some cases of Lennox-Gastaut syndrome. Also found in other syndromes, including genetic epilepsy with febrile seizures plus (GEFS+); autosomal dominant inheritance; presents with febrile seizures at median 1 year, which may persist >6 years, then variable seizure types not associated with fever | Incidence of Dravet syndrome is 1 in 20,000 births, and de novo SCN1A mutation is found in ~80% of cases. Incidence in GEFS+ uncertain; identified in other families with mutations in other sodium channel subunits (SCN2B and SCN2A) and GABA receptor subunit (GABRG2 and GABRA1); significant phenotypic heterogeneity within same family, including members with febrile seizures only. Avoid sodium channel–blocking antiseizure medications |
| LGI1 (10q24) | Leucine-rich glioma-inactivated 1 gene; previous evidence for role in glial tumor progression; recent studies suggest an influence in the postnatal development of glutamatergic circuits in the hippocampus | Autosomal dominant epilepsy with auditory features (ADEAF); a form of lateral temporal lobe epilepsy with auditory symptoms or aphasia as a major focal seizure manifestation; age of onset usually between 10 and 25 years | |
| DEPDC5 (22q12.2) | Disheveled, Egl-10, and pleckstrin domain containing protein 5; exerts an inhibitory effect on mammalian target of rapamycin (mTOR)–mediated processes, such as cell growth and proliferation | Autosomal dominant familial focal epilepsy with variable foci (FFEVF); family members have seizures originating from different cortical regions; neuroimaging usually normal but may harbor subtle malformations; recent studies also suggest association with benign epilepsy with centrotemporal spikes | Study of families with the limited number of affected members revealed mutations in ~12% of families; thus, may be a relatively common cause of lesion-negative focal epilepsies with suspected genetic basis. Also associated with mutations in the GATOR1 genes NPRL2 and NPRL3 |
| GRIN2A (16p13.2) | Encodes NMDA receptor (NMDAR) subunit GluN2A | Spectrum of phenotypes ranging from benign childhood epilepsy with centrotemporal spikes (BECTS) to epilepsy-aphasia syndromes such as Landau-Kleffner syndrome (LKS) and other epileptic encephalopathies | Complex inheritance is implicated, and studies have shown considerable inter- and intrafamilial phenotypic variability and incomplete penetrance |
| CDKL-5 (Xp22.13) | Encodes cyclin-dependent kinase-like 5 (CDKL-5), a serine-threonine kinase involved in neural maturation and synaptogenesis | CDKL-5 deficiency disorder (CDD) results from pathogenic mutation in the CDKL5 gene that causes absence or nonfunctional CDKL-5 protein. CDD is a severe developmental epileptic encephalopathy characterized by very-early-onset seizures. X-linked, affects females more than males | Ganaxolone is a recently approved antiseizure drug that has been shown to significantly reduce CDD-associated seizures |
| SLC2A1 (1p34.2) | Glucose transporter protein type 1 (GLUT1); transports glucose across the blood-brain barrier including in the brain | Loss of function of one allele leads to GLUT1 deficiency, a severe metabolic encephalopathy may respond to standard antiseizure medications, but the gold standard treatment for refractory epilepsy is the ketogenic diet, which bypasses defective glucose transport to provide an alternative energy supply to the brain | Milder forms of epilepsy due to GLUT1 deficiency causes a combination of defective glucose transport, movement disorder (paroxysmal exertional dyskinesia) and epilepsy with prominent absence seizures, though intellect is often normal |
| CSTB (21q22.3) | Cystatin B, a noncaspase cysteine protease inhibitor; normal protein may block neuronal apoptosis by inhibiting caspases directly or indirectly (via cathepsins), or controlling proteolysis | Progressive myoclonus epilepsy (PME) (Unverricht-Lundborg disease); autosomal recessive inheritance; age of onset between 6 and 15 years, myoclonic seizures, ataxia, and progressive cognitive decline; brain shows degeneration associated with polyglucosan intracellular inclusion bodies in numerous organs | Overall rare, but relatively common in Finland and western Mediterranean (>1 in 20,000); precise role of cystatin B in human disease unknown, although mice with null mutations of cystatin B have similar syndrome |
| EPM2A (6q24) | Laforin, a protein tyrosine phosphatase (PTP); involved in glycogen metabolism and may have antiapoptotic activity | Progressive myoclonus epilepsy (Lafora’s disease); autosomal recessive inheritance; age of onset 6–19 years, death within 10 years; brain shows degeneration associated with polyglucosan intracellular inclusion bodies in numerous organs | Most common PME in southern Europe, Middle East, northern Africa, and Indian subcontinent; genetic heterogeneity; unknown whether seizure phenotype due to degeneration or direct effects of abnormal laforin expression |
| Doublecortin (Xq21-24) | Doublecortin, expressed primarily in frontal lobes; directly regulates microtubule polymerization and subcortical band heterotopia | Classic lissencephaly associated with severe mental retardation and seizures in males; increased ascertainment due to improved imaging findings in females (presumably due to random X inactivation); relationship between migration defect and seizure phenotype unknown | Relatively rare but of uncertain incidence; recent studies also suggest association with benign epilepsy with centrotemporal spikes |
4. CLINICAL FEATURES¶
- Focal seizures can have motor manifestations (such as tonic, clonic, or myoclonic movements) or nonmotor manifestations (such as sensory, autonomic, or emotional symptoms) without impairment of awareness.
- A patient having a focal motor seizure arising from the right primary motor cortex near the area controlling hand movement will experience involuntary movements of the contralateral left hand.
- Since the cortical region controlling hand movement is immediately adjacent to the region for facial expression, the seizure may also cause abnormal movements of the face synchronous with the movements of the hand.
- The EEG recorded with scalp electrodes during the seizure may show abnormal focal discharges over the corresponding area of cerebral cortex.
- Focal seizures may also be accompanied by a transient impairment of the patient’s ability to maintain normal contact with the environment.
- The patient is unable to respond appropriately to visual or verbal commands during the seizure and has impaired recollection or awareness of the ictal phase.
- The seizures frequently begin with an aura (i.e., a focal seizure without cognitive disturbance) that is stereotypic for the patient.
- The start of the ictal phase is often a motionless stare, which marks the onset of the period of impaired awareness.
- The impaired awareness may be accompanied by automatisms, involuntary movements that can involve basic behaviors, such as chewing, lip smacking, swallowing, or 'picking' hand movements, or more elaborate behaviors, such as a display of emotion or running.
- The patient is typically disoriented following the seizure, and the transition to full recovery of consciousness may range from seconds to hours.
- Examination immediately following the seizure may show an anterograde amnesia or transient neurologic deficits (such as aphasia, hemi-neglect, or visual loss) caused by postictal inhibition of the cortical regions most involved in the seizure.
- Focal seizures can also evolve into generalized seizures.
- Generalized seizures arise at some point in the brain but immediately and rapidly engage neuronal networks in both cerebral hemispheres.
- Typical absence seizures are characterized by sudden, brief lapses of consciousness without loss of postural control.
- The seizure usually lasts for only seconds, consciousness returns as suddenly as it was lost, and there is no postictal confusion.
- The EEG during the tonic phase of the seizure shows a progressive increase in generalized low-voltage fast activity, followed by generalized high-amplitude, polyspike discharges.
- In the clonic phase, the high-amplitude activity is typically interrupted by slow waves to create a spike-and-slow-wave pattern.
- Generalized seizures tend to terminate synchronously over widespread brain regions.
- The postictal EEG shows diffuse suppression of all cerebral activity, followed by slowing that gradually recovers as the patient awakens.
- Atonic seizures are characterized by sudden loss of postural muscle tone lasting 1–2 s.
- Consciousness is briefly impaired, but there is usually no postictal confusion.
- A very brief seizure may cause only a quick head drop or nodding movement, whereas a longer seizure will cause the patient to collapse (hence, the less formal term, drop attacks).
- Myoclonic seizures are characterized by a sudden and brief muscle contraction that may involve one part of the body or the entire body.
- Pathologic myoclonus is most often seen in association with metabolic disorders, degenerative CNS diseases, or anoxic brain injury.
- Myoclonic seizures are true epileptic events because they are caused by cortical (vs subcortical or spinal) dysfunction.
- The EEG shows bilaterally synchronous spike-and-slow-wave discharges immediately prior to the movement and muscle artifact associated with the myoclonus.
- Myoclonic seizures usually coexist with other forms of generalized seizures but are the predominant feature of juvenile myoclonic epilepsy (JME).
- Epileptic spasms are characterized by a briefly sustained flexion or extension of predominantly proximal muscles, including truncal muscles.
- The EEG usually shows hypsarrhythmia, a chaotic background of diffuse, large-amplitude slow waves and irregular, multifocal spikes and sharp waves.
- During the clinical spasm, there is a marked suppression of the EEG background (the 'electrodecremental response').
- The electromyogram (EMG) also reveals a characteristic rhomboid pattern that may help distinguish spasms from brief tonic and myoclonic seizures.
- Epileptic spasms occur predominantly in infants and likely result from differences in neuronal function and connectivity in the immature versus mature CNS.
4.1 Focal Seizures with Intact Awareness¶
- Focal seizures can have motor manifestations (such as tonic, clonic, or myoclonic movements) or nonmotor manifestations (such as sensory, autonomic, or emotional symptoms) without impairment of awareness.
- Three additional features of focal motor seizures are worth noting:
- First, in some patients, the abnormal motor movements may begin in a very restricted region, such as the fingers, and gradually progress (over seconds to minutes) to include a larger portion of the extremity. This phenomenon, described by Hughlings Jackson and known as a 'Jacksonian march,' represents the spread of seizure activity over a progressively larger region of motor cortex.
- Second, patients may experience a localized paresis (Todd's paralysis) for minutes to many hours in the involved region following the seizure.
- Third, in rare instances, the seizure may continue for hours or days. This condition, termed epilepsia partialis continua, is often refractory to medical therapy.
- Focal seizures may also manifest as changes in somatic sensation (e.g., paresthesias), vision (flashing lights or formed hallucinations), equilibrium (sensation of falling or vertigo), or autonomic function (flushing, sweating, piloerection).
- Focal seizures arising from the temporal lobe may also cause the sensation of acrid odors (e.g., bleach or burning rubber) or tastes (e.g., bitter or metallic), or hearing sounds (simple noise or complex sounds), or an epigastric sensation that rises to the head.
- Some patients describe intense internal feelings such as fear, a dreamlike sense, depersonalization, déjá vu, or illusions that objects are growing smaller (micropsia) or larger (macropsia). These subjective, 'experiential' events that are not directly observable by someone else are referred to as auras.
4.2 Focal Seizures with Impaired Awareness¶
- Focal seizures may also be accompanied by a transient impairment of the patient’s ability to maintain normal contact with the environment.
- The patient is unable to respond appropriately to visual or verbal commands during the seizure and has impaired recollection or awareness of the ictal phase.
- The seizures frequently begin with an aura (i.e., a focal seizure without cognitive disturbance) that is stereotypic for the patient.
- The start of the ictal phase is often a motionless stare, which marks the onset of the period of impaired awareness.
- The impaired awareness may be accompanied by automatisms, involuntary movements that can involve basic behaviors, such as chewing, lip smacking, swallowing, or 'picking' hand movements, or more elaborate behaviors, such as a display of emotion or running.
- The patient is typically disoriented following the seizure, and the transition to full recovery of consciousness may range from seconds to hours.
- Examination immediately following the seizure may show an anterograde amnesia or transient neurologic deficits (such as aphasia, hemi-neglect, or visual loss) caused by postictal inhibition of the cortical regions most involved in the seizure.
4.3 Generalized Onset Seizures¶
- Generalized seizures arise at some point in the brain but immediately and rapidly engage neuronal networks in both cerebral hemispheres.
- Several types of generalized seizures have features that place them in distinctive categories and facilitate clinical diagnosis.
- Typical absence seizures are characterized by sudden, brief lapses of consciousness without loss of postural control.
- The seizure usually lasts for only seconds, consciousness returns as suddenly as it was lost, and there is no postictal confusion.
- Although the brief loss of consciousness may be clinically inapparent or the sole manifestation of the seizure, absence seizures are usually accompanied by subtle, bilateral motor signs such as rapid blinking, chewing movements, or small-amplitude, clonic movements of the hands.
- Typical absence seizures are associated with a group of genetically determined epilepsies with onset usually in childhood (ages 4–10 years) or early adolescence and are the main seizure type in 15–20% of children with epilepsy.
- The seizures can occur hundreds of times per day, but the child may be unaware of or unable to convey their existence.
- Because the clinical signs of the seizures are subtle, especially to parents who may not have had previous experience with seizures, it is not surprising that the first clue to absence epilepsy is often unexplained 'daydreaming' and a decline in school performance recognized by a teacher.
- Indeed, absence epilepsy is often misdiagnosed as an attention deficit disorder.
- Atypical absence seizures have features that deviate both clinically and electrophysiologically from normal, typical absence seizures.
- For example, the lapse of consciousness is usually longer and less abrupt in onset and cessation, and the seizure is accompanied by more obvious motor signs that may include focal or lateralizing features.
- The EEG shows a generalized, slow spike-and-slow-wave pattern with a frequency of ≤2.5 Hz, as well as other abnormal activity.
- Atypical absence seizures are usually associated with diffuse or multifocal structural abnormalities of the brain and therefore may accompany other signs of neurologic dysfunction, such as developmental delay.
- Furthermore, the seizures are less responsive to anticonvulsants compared to typical absence seizures.
- Generalized, tonic-clonic seizures are the main seizure type in ~10% of all persons with epilepsy.
- They are also the most common seizure type resulting from metabolic derangements and are therefore frequently encountered in many different clinical settings.
- The seizure usually begins abruptly without warning, although some patients describe vague premonitory symptoms in the hours leading up to the seizure.
- This prodrome is distinct from the stereotypic auras associated with focal seizures that generalize.
- The initial phase of the seizure is usually tonic contraction of muscles throughout the body, accounting for several classic features of the event.
- Tonic contraction of the muscles of expiration and the larynx at the onset will produce a loud moan or 'ictal cry.'
- Respirations are impaired, secretions pool in the oropharynx, and cyanosis develops.
- Contraction of the jaw muscles may cause biting of the tongue.
- A marked enhancement of sympathetic tone leads to increases in heart rate, blood pressure, and pupillary size.
- After 10–20 s, the tonic phase of the seizure typically evolves into the clonic phase, produced by the superimposition of periods of muscle relaxation on the tonic muscle contraction.
- The periods of relaxation progressively increase until the end of the ictal phase, which usually lasts no more than 1 min.
- The postictal phase is characterized by unresponsiveness, muscular flaccidity, and excessive salivation that can cause stridorous breathing and partial airway obstruction.
- Bladder or bowel incontinence may occur at this point.
- Patients gradually regain consciousness over minutes to hours, and, during this transition, there is typically a period of postictal confusion.
- Patients subsequently complain of headache, fatigue, and muscle ache that can last for many hours.
- The duration of impaired consciousness in the postictal phase can be extremely long (i.e., many hours) in patients with prolonged seizures or underlying central nervous system (CNS) disease.
- Atonic seizures are characterized by sudden loss of postural muscle tone lasting 1–2 s.
- Consciousness is briefly impaired, but there is usually no postictal confusion.
- A very brief seizure may cause only a quick head drop or nodding movement, whereas a longer seizure will cause the patient to collapse (hence, the less formal term, drop attacks).
- This can be extremely dangerous because there is a substantial risk of direct head injury with the fall.
- The EEG shows brief, generalized spike-and-wave discharges followed immediately by diffuse slow waves that correlate with the loss of muscle tone.
- Like pure tonic seizures, atonic seizures are usually seen in association with known epilepsy syndromes.
- Myoclonic seizures are characterized by a sudden and brief muscle contraction that may involve one part of the body or the entire body.
- A common physiologic form of myoclonus is the sudden jerking movement observed while falling asleep.
- Pathologic myoclonus is most often seen in association with metabolic disorders, degenerative CNS diseases, or anoxic brain injury.
- Although the distinction from other forms of myoclonus is imprecise, myoclonic seizures are true epileptic events because they are caused by cortical (vs subcortical or spinal) dysfunction.
- The EEG shows bilaterally synchronous spike-and-slow-wave discharges immediately prior to the movement and muscle artifact associated with the myoclonus.
- Myoclonic seizures usually coexist with other forms of generalized seizures but are the predominant feature of juvenile myoclonic epilepsy (JME).
- Epileptic spasms are characterized by a briefly sustained flexion or extension of predominantly proximal muscles, including truncal muscles.
- The EEG usually shows hypsarrhythmia, a chaotic background of diffuse, large-amplitude slow waves and irregular, multifocal spikes and sharp waves.
- During the clinical spasm, there is a marked suppression of the EEG background (the 'electrodecremental response').
- The electromyogram (EMG) also reveals a characteristic rhomboid pattern that may help distinguish spasms from brief tonic and myoclonic seizures.
- Epileptic spasms occur predominantly in infants and likely result from differences in neuronal function and connectivity in the immature versus mature CNS.
5. DIFFERENTIAL DIAGNOSIS¶
- Extreme caution is advised before concluding that paroxysmal, stereotypic episodes of bizarre or atypical behavior are not due to seizure activity.
- In such cases, additional detailed EEG studies may be helpful.
- Distinguishing between focal and generalized onset seizures is extremely important, because there are substantial differences in the evaluation and treatment of epilepsies characterized by focal versus generalized onset seizures.
- Bystanders tend to emphasize the more dramatic, generalized convulsive phase of the seizure and overlook the more subtle, focal symptoms present at onset.
- In some cases, the focal onset of the seizure becomes apparent only when a careful history identifies a preceding aura.
- Often, however, the focal onset is not clinically evident and may be established only through careful EEG analysis.
6. INVESTIGATIONS & DIAGNOSIS¶
- The routine interictal (i.e., between seizures) electroencephalogram (EEG) in patients with focal seizures is often normal or may show brief discharges termed epileptiform spikes, or sharp waves.
- Because focal seizures can arise from the medial temporal lobe or inferior frontal lobe (i.e., regions distant from the scalp), the EEG recorded during the seizure may be nonlocalizing.
- However, the region of seizure onset may be defined using surgically placed intracranial electrodes.
- The electrophysiologic hallmark of typical absence seizures is a burst of generalized, symmetric, 3-Hz, spike-and-slow-wave discharges that begins and ends suddenly, superimposed on a normal EEG background.
- Periods of spike-and-slow-wave discharges lasting more than a few seconds usually correlate with clinical signs, but the EEG often shows many more brief bursts of abnormal cortical activity than suspected clinically.
- Hyperventilation tends to provoke these electrographic discharges and even the seizures themselves and is routinely used when recording the EEG.
- The EEG during the tonic phase of the seizure shows a progressive increase in generalized low-voltage fast activity, followed by generalized high-amplitude, polyspike discharges.
- In the clonic phase, the high-amplitude activity is typically interrupted by slow waves to create a spike-and-slow-wave pattern.
- Generalized seizures tend to terminate synchronously over widespread brain regions.
- The postictal EEG shows diffuse suppression of all cerebral activity, followed by slowing that gradually recovers as the patient awakens.
- High-resolution magnetic resonance imaging (MRI) can detect the characteristic hippocampal sclerosis that appears to be essential in the pathophysiology of MTLE for many patients.
- Recognition of this syndrome is especially important because it tends to be refractory to treatment with anticonvulsants but responds well to surgical intervention.
6.1 Diagnostic Criteria and Algorithms¶
- Determining the type of seizure that has occurred is essential for focusing the diagnostic approach on particular etiologies, selecting appropriate therapy, and providing information regarding prognosis.
- The International League Against Epilepsy (ILAE) Commission on Classification and Terminology updated their approach to classification of seizures in 2017.
- This system is based on the clinical features of seizures and associated electroencephalographic findings.
- Other potentially distinctive features such as etiology or cellular substrate are not considered in this classification system, although this will undoubtedly change in the future as more is learned about the pathophysiologic mechanisms that underlie specific seizure types.
- A fundamental principle is that seizures may be either focal or generalized.
- Focal seizures originate within networks limited to one brain region.
- Generalized seizures arise within and rapidly engage networks distributed across both cerebral hemispheres.
- Focal seizures are often associated with structural abnormalities of the brain.
- In contrast, generalized seizures may result from cellular, biochemical, or structural abnormalities with a more widespread distribution.
- There are exceptions in both cases, however.
- Using the definition of epilepsy as two or more unprovoked seizures, the incidence of epilepsy is ~0.3–0.5% in different populations throughout the world.
- The prevalence of epilepsy has been estimated at 5–30 persons per 1000.
- ~5–10% of the population will have at least one seizure, with the highest incidence occurring in early childhood and late adulthood.
- The meaning of the term seizure needs to be carefully distinguished from that of epilepsy.
- Epilepsy describes a condition in which a person has a risk of recurrent seizures due to a chronic, underlying process.
- This definition implies that a person with a single seizure, or recurrent seizures due to correctable or avoidable circumstances, does not necessarily have epilepsy.
- A single seizure associated with clinical or electroencephalographic features portending high risk of recurrence may establish the diagnosis of epilepsy.
- Epilepsy refers to a clinical phenomenon rather than a single disease entity because many forms and causes exist.
- However, among the many causes of epilepsy, there are various epilepsy syndromes in which the clinical and pathologic characteristics are distinctive and suggest a specific underlying etiology.
7. MANAGEMENT & TREATMENT¶
- Seizures are episodic and occur intermittently.
- Depending on the underlying cause, people with epilepsy may feel completely normal for months or years between seizures.
- This implies there are important provocative or precipitating factors that induce seizures in people with epilepsy.
- Similarly, precipitating factors are responsible for causing the single seizure in someone without epilepsy.
- Precipitants include those due to intrinsic physiologic processes, such as psychological or physical stress, sleep deprivation, or hormonal changes.
- They also include exogenous factors such as exposure to toxic substances, certain medications, and intermittent photic stimulation from strobe lights or some video games.
- These observations emphasize the concept that the many causes of seizures and epilepsy result from a dynamic interplay between endogenous factors, epileptogenic factors, and precipitating factors.
- The potential role of each needs to be considered when determining the appropriate management of a patient with seizures.
- For example, the identification of predisposing factors (e.g., family history of epilepsy) in a patient with febrile seizures may increase the necessity for closer follow-up and a more aggressive diagnostic evaluation.
- Finding an epileptogenic lesion may help in the estimation of seizure recurrence and duration of therapy.
- Removal or modification of a precipitating factor may be an effective and safer method for preventing further seizures than the prophylactic use of anticonvulsant drugs.
- An emerging concept holds that underlying seizure risk itself fluctuates cyclically, potentially explaining why the same precipitating factor (e.g., sleep deprivation) can be well tolerated on some occasions but result in a seizure on others.
- Ganaxolone is a recently approved antiseizure drug that has been shown to significantly reduce CDD-associated seizures.
- The ketogenic diet is the gold standard treatment for refractory epilepsy due to GLUT1 deficiency.
- Avoid sodium channel–blocking antiseizure medications for SCN1A-related epilepsy (Dravet syndrome).
- Advances in the understanding of basic mechanisms of epilepsy have come through studies of experimental models of MTLE.
- Implanted neurostimulation devices are now being investigated for treatment of seizures in Lennox-Gastaut syndrome and other generalized epilepsies.
- An allogeneic bone marrow–derived MSC line received conditional marketing approval in Japan in 2024 for the indication of improving chronic motor paralysis resulting from traumatic brain injury.
- The MSCs were transiently transfecting with the human Notch-1 intracellular domain gene to promote FDF-2 secretion in order to 'enhance their ability to regenerate nerve cells' according to the pharmaceutical company that developed the cell-based therapy.
- The approval followed results of a phase 2 clinical trial conducted in Japan and the United States.
- Forty-six patients with moderate to severe traumatic brain injury and chronic motor deficits had MSCs stereotactically infused into an area of encephalomalacia identified on MRI scan while a sham group of 15 patients had burr holes only.
- The trial met the primary endpoint showing significant improvement in motor function at 24 weeks on the Fugle-Meyer Motor Scale (FMMS) (p = .04).
- Interestingly, a small improvement was noted in the sham-treated group as well, indicating the presence of a placebo effect.
- A larger, double-blind, randomized, sham-controlled study is now planned.
7.1 Pharmacologic and Non-Pharmacologic Therapies¶
- Seizures are a result of a shift in the normal balance of excitation and inhibition within the CNS.
- Given the numerous properties that control neuronal excitability, it is not surprising that there are many ways to perturb this normal balance and, therefore, many different causes of both seizures and epilepsy.
- Three clinical observations emphasize how a variety of factors determine why certain conditions may cause seizures or epilepsy in a given patient.
-
- The normal brain can have a seizure under the appropriate circumstances, and there are differences between individuals in the susceptibility or threshold for seizures.
-
- There are a variety of conditions that have an extremely high likelihood of resulting in a chronic seizure disorder. One of the best examples of this is severe, penetrating head trauma, which is associated with up to a 45% risk of subsequent epilepsy.
-
- Seizures are episodic.
- Precipitants include those due to intrinsic physiologic processes, such as psychological or physical stress, sleep deprivation, or hormonal changes.
- They also include exogenous factors such as exposure to toxic substances, certain medications, and intermittent photic stimulation from strobe lights or some video games.
- These observations emphasize the concept that the many causes of seizures and epilepsy result from a dynamic interplay between endogenous factors, epileptogenic factors, and precipitating factors.
- The potential role of each needs to be considered when determining the appropriate management of a patient with seizures.
- For example, the identification of predisposing factors (e.g., family history of epilepsy) in a patient with febrile seizures may increase the necessity for closer follow-up and a more aggressive diagnostic evaluation.
- Finding an epileptogenic lesion may help in the estimation of seizure recurrence and duration of therapy.
- Removal or modification of a precipitating factor may be an effective and safer method for preventing further seizures than the prophylactic use of anticonvulsant drugs.
- An emerging concept holds that underlying seizure risk itself fluctuates cyclically, potentially explaining why the same precipitating factor (e.g., sleep deprivation) can be well tolerated on some occasions but result in a seizure on others.
- Ganaxolone is a recently approved antiseizure drug that has been shown to significantly reduce CDD-associated seizures.
- The ketogenic diet is the gold standard treatment for refractory epilepsy due to GLUT1 deficiency.
- Avoid sodium channel–blocking antiseizure medications for SCN1A-related epilepsy (Dravet syndrome).
- Advances in the understanding of basic mechanisms of epilepsy have come through studies of experimental models of MTLE.
- Implanted neurostimulation devices are now being investigated for treatment of seizures in Lennox-Gastaut syndrome and other generalized epilepsies.
- An allogeneic bone marrow–derived MSC line received conditional marketing approval in Japan in 2024 for the indication of improving chronic motor paralysis resulting from traumatic brain injury.
- The MSCs were transiently transfecting with the human Notch-1 intracellular domain gene to promote FDF-2 secretion in order to 'enhance their ability to regenerate nerve cells' according to the pharmaceutical company that developed the cell-based therapy.
- The approval followed results of a phase 2 clinical trial conducted in Japan and the United States.
- Forty-six patients with moderate to severe traumatic brain injury and chronic motor deficits had MSCs stereotactically infused into an area of encephalomalacia identified on MRI scan while a sham group of 15 patients had burr holes only.
- The trial met the primary endpoint showing significant improvement in motor function at 24 weeks on the Fugle-Meyer Motor Scale (FMMS) (p = .04).
- Interestingly, a small improvement was noted in the sham-treated group as well, indicating the presence of a placebo effect.
- A larger, double-blind, randomized, sham-controlled study is now planned.
8. PROGNOSIS & COMPLICATIONS¶
- Although complete remission is uncommon, the seizures usually respond well to appropriate antiseizure medication.
- There is often a family history of epilepsy, and genetic studies suggest a polygenic cause.
- Lennox-Gastaut syndrome is associated with CNS disease or dysfunction from a variety of causes, including de novo mutations, developmental abnormalities, perinatal hypoxia/ischemia, trauma, infection, and other acquired lesions.
- The multifactorial nature of this syndrome suggests that it is a nonspecific response of the brain to diffuse neuronal dysfunction.
- Unfortunately, many patients have a poor prognosis due to the underlying CNS disease and the physical and psychosocial consequences of severe, poorly controlled epilepsy.
- Severe, penetrating head trauma is associated with up to a 45% risk of subsequent epilepsy.
- The high propensity for severe traumatic brain injury to lead to epilepsy suggests that the injury results in a long-lasting pathologic change in the CNS that transforms a presumably normal neuronal network into one that is abnormally hyperexcitable.
- This process is known as epileptogenesis, and the specific changes that result in a lowered seizure threshold can be considered epileptogenic factors.
- Other processes associated with epileptogenesis include stroke, infection, neurodegeneration, and abnormalities of CNS development.
- Seizures are episodic and occur intermittently.
- Depending on the underlying cause, people with epilepsy may feel completely normal for months or years between seizures.
- This implies there are important provocative or precipitating factors that induce seizures in people with epilepsy.
- Similarly, precipitating factors are responsible for causing the single seizure in someone without epilepsy.
- Precipitants include those due to intrinsic physiologic processes, such as psychological or physical stress, sleep deprivation, or hormonal changes.
- They also include exogenous factors such as exposure to toxic substances, certain medications, and intermittent photic stimulation from strobe lights or some video games.
- These observations emphasize the concept that the many causes of seizures and epilepsy result from a dynamic interplay between endogenous factors, epileptogenic factors, and precipitating factors.
- The potential role of each needs to be considered when determining the appropriate management of a patient with seizures.
- For example, the identification of predisposing factors (e.g., family history of epilepsy) in a patient with febrile seizures may increase the necessity for closer follow-up and a more aggressive diagnostic evaluation.
- Finding an epileptogenic lesion may help in the estimation of seizure recurrence and duration of therapy.
- Removal or modification of a precipitating factor may be an effective and safer method for preventing further seizures than the prophylactic use of anticonvulsant drugs.
- An emerging concept holds that underlying seizure risk itself fluctuates cyclically, potentially explaining why the same precipitating factor (e.g., sleep deprivation) can be well tolerated on some occasions but result in a seizure on others.
- The duration of impaired consciousness in the postictal phase can be extremely long (i.e., many hours) in patients with prolonged seizures or underlying central nervous system (CNS) disease.
9. SPECIAL CONSIDERATIONS¶
- In practice, it is useful to consider the etiologies of seizures based on the age of the patient, because age is one of the most important factors in determining both the incidence and the likely causes of seizures or epilepsy.
- During the neonatal period and early infancy, potential causes include hypoxic-ischemic encephalopathy, trauma, CNS infection, congenital CNS abnormalities, and metabolic disorders.
- Babies born to mothers using neurotoxic drugs such as cocaine, heroin, or ethanol are susceptible to drug-withdrawal seizures in the first few days after delivery.
- Hypoglycemia and hypocalcemia, which can occur as secondary complications of perinatal injury, are also causes of early postnatal seizures.
- Seizures due to inborn errors of metabolism usually present once regular feeding begins, typically 2–3 days after birth.
- Pyridoxine (vitamin B6) deficiency, an important cause of neonatal seizures, is treated with pyridoxine replacement.
- Idiopathic or familial forms of benign neonatal seizures are also seen during this time.
- The most common seizures arising in late infancy and early childhood are febrile seizures, which are seizures associated with fevers but without evidence of CNS infection or other defined causes.
- The overall prevalence is 3–5% and even higher in some parts of the world such as Asia.
- Patients often have a family history of febrile seizures or epilepsy.
- Febrile seizures usually occur between 3 months and 5 years of age and have a peak incidence between 18 and 24 months.
- The typical scenario is a child who has a generalized, tonic-clonic seizure during a febrile illness in the setting of a common childhood infection such as otitis media.
- Typical absence seizures are associated with a group of genetically determined epilepsies with onset usually in childhood (ages 4–10 years) or early adolescence and are the main seizure type in 15–20% of children with epilepsy.
- The seizures can occur hundreds of times per day, but the child may be unaware of or unable to convey their existence.
- Because the clinical signs of the seizures are subtle, especially to parents who may not have had previous experience with seizures, it is not surprising that the first clue to absence epilepsy is often unexplained 'daydreaming' and a decline in school performance recognized by a teacher.
- Indeed, absence epilepsy is often misdiagnosed as an attention deficit disorder.
- Atypical absence seizures are usually associated with diffuse or multifocal structural abnormalities of the brain and therefore may accompany other signs of neurologic dysfunction, such as developmental delay.
- Furthermore, the seizures are less responsive to anticonvulsants compared to typical absence seizures.
9.1 Age-Related Etiologies¶
- In practice, it is useful to consider the etiologies of seizures based on the age of the patient, because age is one of the most important factors in determining both the incidence and the likely causes of seizures or epilepsy.
- During the neonatal period and early infancy, potential causes include hypoxic-ischemic encephalopathy, trauma, CNS infection, congenital CNS abnormalities, and metabolic disorders.
- Babies born to mothers using neurotoxic drugs such as cocaine, heroin, or ethanol are susceptible to drug-withdrawal seizures in the first few days after delivery.
- Hypoglycemia and hypocalcemia, which can occur as secondary complications of perinatal injury, are also causes of early postnatal seizures.
- Seizures due to inborn errors of metabolism usually present once regular feeding begins, typically 2–3 days after birth.
- Pyridoxine (vitamin B6) deficiency, an important cause of neonatal seizures, is treated with pyridoxine replacement.
- Idiopathic or familial forms of benign neonatal seizures are also seen during this time.
- The most common seizures arising in late infancy and early childhood are febrile seizures, which are seizures associated with fevers but without evidence of CNS infection or other defined causes.
- The overall prevalence is 3–5% and even higher in some parts of the world such as Asia.
- Patients often have a family history of febrile seizures or epilepsy.
- Febrile seizures usually occur between 3 months and 5 years of age and have a peak incidence between 18 and 24 months.
- The typical scenario is a child who has a generalized, tonic-clonic seizure during a febrile illness in the setting of a common childhood infection such as otitis media.
10. KEY PEARLS & CLINICAL TRAPS¶
- The meaning of the term seizure needs to be carefully distinguished from that of epilepsy.
- Epilepsy describes a condition in which a person has a risk of recurrent seizures due to a chronic, underlying process.
- This definition implies that a person with a single seizure, or recurrent seizures due to correctable or avoidable circumstances, does not necessarily have epilepsy.
- A single seizure associated with clinical or electroencephalographic features portending high risk of recurrence may establish the diagnosis of epilepsy.
- Epilepsy refers to a clinical phenomenon rather than a single disease entity because many forms and causes exist.
- However, among the many causes of epilepsy, there are various epilepsy syndromes in which the clinical and pathologic characteristics are distinctive and suggest a specific underlying etiology.
- Extreme caution is advised before concluding that paroxysmal, stereotypic episodes of bizarre or atypical behavior are not due to seizure activity.
- In such cases, additional detailed EEG studies may be helpful.
- Distinguishing between focal and generalized onset seizures is extremely important, because there are substantial differences in the evaluation and treatment of epilepsies characterized by focal versus generalized onset seizures.
- Bystanders tend to emphasize the more dramatic, generalized convulsive phase of the seizure and overlook the more subtle, focal symptoms present at onset.
- In some cases, the focal onset of the seizure becomes apparent only when a careful history identifies a preceding aura.
- Often, however, the focal onset is not clinically evident and may be established only through careful EEG analysis.
- The electrophysiologic hallmark of typical absence seizures is a burst of generalized, symmetric, 3-Hz, spike-and-slow-wave discharges that begins and ends suddenly, superimposed on a normal EEG background.
- Hyperventilation tends to provoke these electrographic discharges and even the seizures themselves and is routinely used when recording the EEG.
- Avoid sodium channel–blocking antiseizure medications for SCN1A-related epilepsy (Dravet syndrome).
- The ketogenic diet is the gold standard treatment for refractory epilepsy due to GLUT1 deficiency.
- Ganaxolone is a recently approved antiseizure drug that has been shown to significantly reduce CDD-associated seizures.
- Severe, penetrating head trauma is associated with up to a 45% risk of subsequent epilepsy.
- The high propensity for severe traumatic brain injury to lead to epilepsy suggests that the injury results in a long-lasting pathologic change in the CNS that transforms a presumably normal neuronal network into one that is abnormally hyperexcitable.
- This process is known as epileptogenesis, and the specific changes that result in a lowered seizure threshold can be considered epileptogenic factors.
- Other processes associated with epileptogenesis include stroke, infection, neurodegeneration, and abnormalities of CNS development.
- Seizures are episodic and occur intermittently.
- Depending on the underlying cause, people with epilepsy may feel completely normal for months or years between seizures.
- This implies there are important provocative or precipitating factors that induce seizures in people with epilepsy.
- Similarly, precipitating factors are responsible for causing the single seizure in someone without epilepsy.
- Precipitants include those due to intrinsic physiologic processes, such as psychological or physical stress, sleep deprivation, or hormonal changes.
- They also include exogenous factors such as exposure to toxic substances, certain medications, and intermittent photic stimulation from strobe lights or some video games.
- These observations emphasize the concept that the many causes of seizures and epilepsy result from a dynamic interplay between endogenous factors, epileptogenic factors, and precipitating factors.
- The potential role of each needs to be considered when determining the appropriate management of a patient with seizures.
- For example, the identification of predisposing factors (e.g., family history of epilepsy) in a patient with febrile seizures may increase the necessity for closer follow-up and a more aggressive diagnostic evaluation.
- Finding an epileptogenic lesion may help in the estimation of seizure recurrence and duration of therapy.
- Removal or modification of a precipitating factor may be an effective and safer method for preventing further seizures than the prophylactic use of anticonvulsant drugs.
- An emerging concept holds that underlying seizure risk itself fluctuates cyclically, potentially explaining why the same precipitating factor (e.g., sleep deprivation) can be well tolerated on some occasions but result in a seizure on others.
Flowcharts & Algorithms¶
Reproduced from Harrison's 22nd Edition.
Flowchart 1¶

Caption: FIGURE 436-2 Evaluation of the adult patient with a seizure. CBC, complete blood count; MRI, magnetic resonance imaging. milestones may provide evidence for underlying CNS disease. Precipi- tating factors such as sleep deprivation, systemic diseases, electrolyte or metabolic derangements, acute infection, drugs that lower the seizure threshold (Table 436-5), or alcohol or illicit drug use should also be identified. The general physical examination includes a search for signs of infection or systemic illness. Careful examination of the skin may
Flowchart 2¶

Caption: FIGURE 436-5 Pharmacologic treatment of generalized tonic-clonic status epilepticus LEV, levetiracetam; LZP, lorazepam; MDZ, midazolam; PGB, pregabalin; PHT, phenytoin or rTMS, repetitive transcranial magnetic stimulation; THP, thiopental; TPM, topiramate; Lowenstein: Management of refractory status epilepticus in adults: still more questions
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶
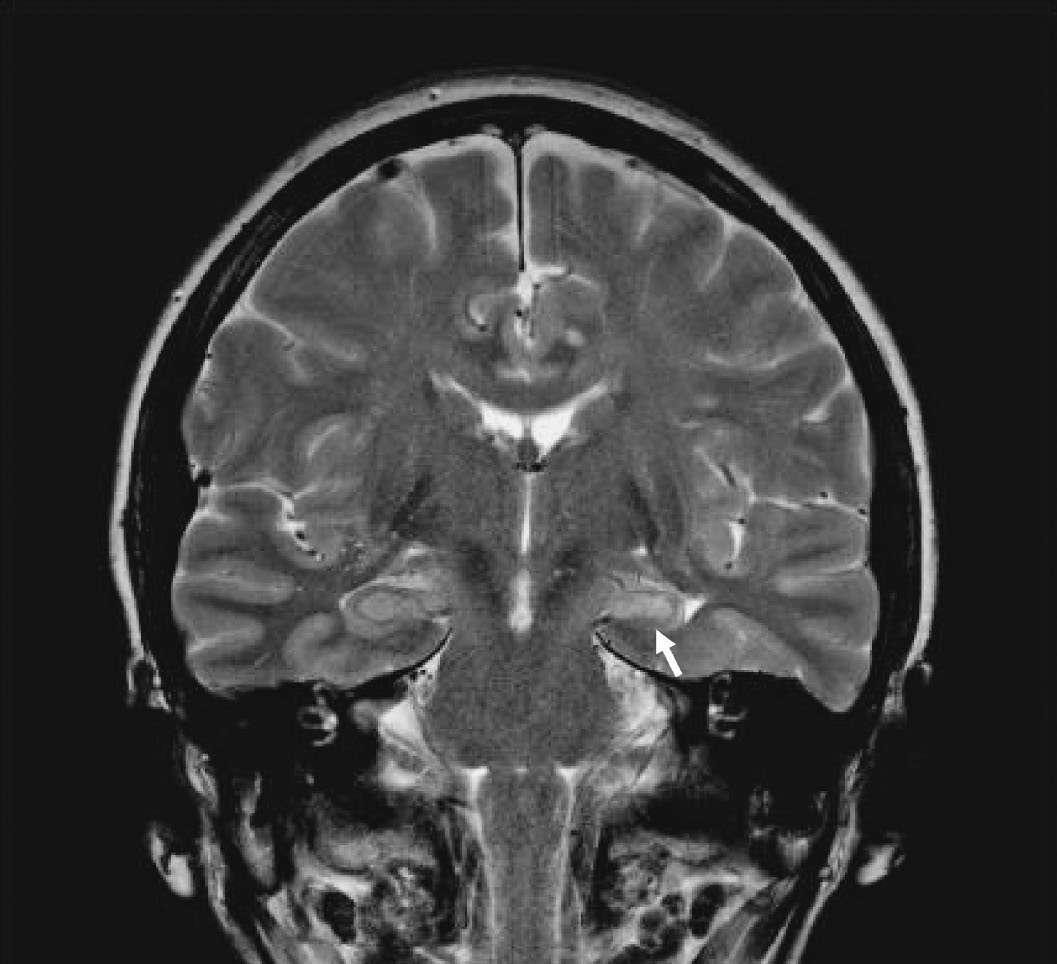
Caption: FIGURE 436-1 Mesial temporal lobe epilepsy. The electroencephalogram and seizure semiology were consistent with a left temporal lobe focus. This coronal high-resolution T2-weighted fast spin echo magnetic resonance image obtained at of 3 Tesla is at the level of the hippocampal bodies and shows abnormal high signal of intensity, blurring of internal laminar architecture, and reduced size of the left hippocampus (arrow) relative to the right. This triad of imaging findings is consistent with hippocampal sclerosis. — Figure 436-1: EEG tracing demonstrating generalized, symmetric 3-Hz spike-and-slow-wave discharges characteristic of typical absence seizures, often provoked by hyperventilation.
Figure 2¶

Caption: FIGURE 436-4 Electrographic seizures. A. Onset of a tonic seizure showing generalized repetitive sharp activity with synchronous onset over both hemispheres. B. Burst of repetitive spikes occurring with sudden onset in the right temporal region during a clinical spell characterized by transient impairment of awareness. C. Generalized 3-Hz spike-wave activity occurring synchronously over both hemispheres during an absence seizure. Horizontal calibration: 1 s; vertical calibration: 400 μV in A, 200 μV in B, and 750 μV in C. (Reproduced with permission from MJ Aminoff: Aminoff’s Electrodiagnosis in Clinical Neurology, 6th ed. Oxford: Elsevier Saunders, 2012.) The routine scalp EEG may also be used to assess the prognosis of — Figure 436-2: High-resolution MRI of the mesial temporal lobe showing hippocampal sclerosis, the characteristic pathologic feature of mesial temporal lobe epilepsy syndrome.
Figure 3¶

Caption: FIGURE 436-3 Electroencephalograms. A. Normal electroencephalogram (EEG) alpha rhythm that attenuates with eye opening. B. Abnormal EEG showing irregular diffuse patient with encephalitis. C. Irregular slow activity in the right central region, on a patient with a right parietal glioma. D. Periodic complexes occurring once every second Jakob disease. Horizontal calibration: 1 s; vertical calibration: 200 μV in A, 300 μV in following figure, electrode placements are indicated at the left of each panel and accord system. A, earlobe; C, central; F, frontal; Fp, frontal polar; O, occipital; P, parietal; T, are indicated by even numbers, left-sided placements by odd numbers, and midline permission from MJ Aminoff: Aminoff’s Electrodiagnosis in Clinical Neurology, 6th ed. — Figure 436-3: Clinical photograph illustrating the tonic-clonic phase of a generalized seizure, showing muscle stiffening followed by rhythmic jerking and loss of consciousness.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.