Peripheral Neuropathy¶
Chapter 457 | Part 13: Neurologic Disorders · Part 13 – Neurologic Disorders
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- Approximately half of patients with generalized symmetric peripheral neuropathy have no identifiable etiology (idiopathic or cryptogenic sensory and sensorimotor polyneuropathy).
- Symmetric proximal and distal weakness with sensory loss is the hallmark of inflammatory demyelinating polyneuropathy (GBS and CIDP).
- CMT1 is the most common form of hereditary neuropathy, characterized by motor conduction velocities <38 m/s and onion bulb formation on biopsy.
- Small-fiber neuropathy is likely if pain and temperature perception are lost while vibratory and position sense are preserved, with normal NCS.
- Vitamin B12 deficiency is the most common cause of combined system degeneration with neuropathy and upper motor neuron involvement.
- Nerve biopsy is rarely performed now; primary indications are suspicion for amyloid neuropathy or vasculitis.
- Skin biopsy can diagnose small-fiber neuropathy by measuring density of small unmyelinated fibers.
- CMT1A is caused by a 1.5-Mb duplication within chromosome 17p11.2-12 encoding the gene for peripheral myelin protein-22 (PMP-22).
- In patients with suspected GBS or CIDP, CSF pleocytosis is usually absent; if cells are present, consider HIV, Lyme, sarcoidosis, or lymphomatous infiltration.
- Autonomic dysfunction in the absence of diabetes should alert the clinician to the possibility of amyloid polyneuropathy.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 General Approach
- 1.2 Anatomy & Classification
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Hereditary Neuropathies
- 3.2 Acquired Neuropathies
- 4. CLINICAL FEATURES
- 4.1 History & Physical Examination
- 4.2 Pattern Recognition
- 5. DIFFERENTIAL DIAGNOSIS
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Electrodiagnostic Studies
- 6.2 Other Laboratory & Imaging
- 6.3 Biopsy
- 7. MANAGEMENT & TREATMENT
- 7.1 Hereditary Neuropathies
- 7.2 Acquired Neuropathies
- 8. PROGNOSIS & COMPLICATIONS
- 9. SPECIAL CONSIDERATIONS
- 10. KEY PEARLS & CLINICAL TRAPS
- Flowcharts & Algorithms
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🔀 Flowchart | Approach to the evaluation of peripheral neuropathies |
| 1 | 🖼 Figure | Lumbosacral plexus |
| 2 | 🖼 Figure | Brachial plexus anatomy |
| 3 | 🖼 Figure | Lumbosacral plexus |
1. DEFINITION & OVERVIEW¶
Peripheral nerves are composed of sensory, motor, and autonomic elements. Diseases can affect the cell body of a neuron or its peripheral processes, namely the axons or the encasing myelin sheaths. Most peripheral nerves are mixed and contain sensory and motor as well as autonomic fibers. Nerves can be subdivided into three major classes: large myelinated, small myelinated, and small unmyelinated. Motor axons are usually large myelinated fibers that conduct rapidly (~50 m/s). Sensory fibers may be any of the three types. Large-diameter sensory fibers conduct proprioception and vibratory sensation to the brain, while the smaller-diameter myelinated and unmyelinated fibers transmit pain and temperature sensation. Autonomic nerves are also small in diameter. Thus, peripheral neuropathies can impair sensory, motor, or autonomic function, either singly or in combination. Peripheral neuropathies are further classified into those that primarily affect the cell body (e.g., neuronopathy or ganglionopathy), myelin (myelinopathy), and the axon (axonopathy). These different classes of peripheral neuropathies have distinct clinical and electrophysiologic features.
1.1 General Approach¶
In approaching a patient with a neuropathy, the clinician has three main goals: (1) identify where the lesion is, (2) identify the cause, and (3) determine the proper treatment. The first goal is accomplished by obtaining a thorough history, neurologic examination, and electrodiagnostic and other laboratory studies. While gathering this information, seven key questions are asked, the answers to which help identify the pattern of involvement and the cause of the neuropathy. Despite an extensive evaluation, in approximately half of patients, no etiology is ever found; these patients typically have a predominately sensory polyneuropathy and have been labeled as having idiopathic or cryptogenic sensory and sensorimotor polyneuropathy (CSPN). The majority of neuropathies are predominantly sensory in nature.
1.2 Anatomy & Classification¶
Peripheral nerves are composed of sensory, motor, and autonomic elements. Diseases can affect the cell body of a neuron or its peripheral processes, namely the axons or the encasing myelin sheaths. Most peripheral nerves are mixed and contain sensory and motor as well as autonomic fibers. Nerves can be subdivided into three major classes: large myelinated, small myelinated, and small unmyelinated. Motor axons are usually large myelinated fibers that conduct rapidly (~50 m/s). Sensory fibers may be any of the three types. Large-diameter sensory fibers conduct proprioception and vibratory sensation to the brain, while the smaller-diameter myelinated and unmyelinated fibers transmit pain and temperature sensation. Autonomic nerves are also small in diameter. Thus, peripheral neuropathies can impair sensory, motor, or autonomic function, either singly or in combination. Peripheral neuropathies are further classified into those that primarily affect the cell body (e.g., neuronopathy or ganglionopathy), myelin (myelinopathy), and the axon (axonopathy). These different classes of peripheral neuropathies have distinct clinical and electrophysiologic features.
2. EPIDEMIOLOGY¶
Approximately half of patients with generalized symmetric peripheral neuropathy have no identifiable etiology. CMT disease is the most common type of hereditary neuropathy. Diabetes mellitus is a common cause of acquired neuropathy. The majority of neuropathies are predominantly sensory in nature.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
Peripheral neuropathies can be caused by hereditary factors, acquired metabolic disorders, drugs, toxins, infections, autoimmune diseases, and paraneoplastic syndromes. Hereditary neuropathies include Charcot-Marie-Tooth disease (CMT). Acquired neuropathies include diabetic neuropathy, amyloidosis, vasculitis, and toxic neuropathies. Pathophysiology involves axonal degeneration or segmental demyelination. Axonal degeneration is characterized by low-amplitude potentials with relatively preserved distal latencies, conduction velocities, and late potentials, along with fibrillations on needle EMG. Segmental demyelination is characterized by slow conduction velocities, prolonged distal latencies, conduction block, and temporal dispersion.
3.1 Hereditary Neuropathies¶
CMT disease is a syndrome of many genetically distinct disorders. The various subtypes of CMT are classified according to the nerve conduction velocities (NCVs) and predominant pathology (e.g., demyelination or axonal degeneration), inheritance pattern (autosomal dominant, autosomal recessive, or X-linked), and the specific mutated genes. Type 1 CMT (or CMT1) refers to inherited demyelinating sensorimotor neuropathies, whereas the axonal sensory neuropathies are classified as CMT2. By definition, motor conduction velocities in the arms are slowed to 38 m/s in CMT2. However, most cases of CMT1 actually have motor NCVs between 20 and 25 m/s. CMT1 and CMT2 usually begin in childhood or early adult life; however, onset later in life can occur, particularly in CMT2. Both are inherited in an autosomal dominant fashion, with a few exceptions. There are no medical therapies for any of the CMTs, but physical and occupational therapy can be beneficial, as can bracing (e.g., ankle-foot orthotics for foot drop) and other orthotic devices.
Table 1 — Table 457-4 Classification of Charcot-Marie-Tooth Disease and Related Neuropathies¶
| NAME | INHERITANCE | GENE LOCATION | GENE |
|---|---|---|---|
| CMT1A | AD | 17p11.2 | PMP22 (usually duplication of gene) |
| CMT1B | AD | 1q21-23 | MPZ |
| CMT1C | AD | 16p13.1-p12.3 | LITAF |
| CMT1D | AD | 10q21.1-22.1 | ERG2 |
| CMT1E (with deafness) | AD | 17p11.2 | PMP22 gene (usually point mutations) |
| CMT1F | AD | 8p13-21 | NEFL |
| CMT1G | AD | 8q21 | PMP22 |
| CMT2A2 (allelic to HMSN VI with optic atrophy) | AD | 1p36.2 | MFN2 |
| CMT2B | AD | 3q13-q22 | RAB7 |
| CMT2B1 (allelic to LGMD 1B) | AR | 1q21.2 | LMNA |
| CMT2B2 | AR | 19q13 | PNKP |
| CMT2C (allelic to scapuloperoneal neuropathy) | AD | 12q23-24 | TRPV4 |
| CMT2D (allelic to distal SMA5) | AD | 7p14 | GARS1 |
| CMT2DD | AD | 1p13 | ATP1A1 |
| CMT2E (allelic to CMT1F) | AD | 8p21 | NEFL |
| CMT2EE | AD | 2p23 | MPV17 |
| CMT2F | AD | 7q11-q21 | HSPB1 |
| CMT2G (allelic to CMT2P) | AD | 9q31.3-34.2 | LRSAM1 |
| CMT2I (allelic to CMT1B) | AD | 1q22 | MPZ |
| CMT2J | AD | 1q22 | MPZ |
| CMT2H, CMT2K (allelic to CMT4A) | AD | 8q13-q21 | GDAP1 |
| CMT2L (allelic to distal hereditary motor neuropathy type 2) | AD | 12q24 | HSPB8 |
| CMT2M | AD | 16q22 | DNM2 |
| CMT2N | AD | 16q22.1 | AARS |
| CMT2O | AD | 14q32.31 | DYNC1H1 |
| CMT2P | AD and AR | 9q31.3-34.2 | LRSAM1 |
| CMT2P-Okinawa (allelic to HSMN2P) | AD | 3q13-q14 | TFG |
| CMT2Q | AD | 10p14 | DHTKD1 |
| CMT2R | AD | 4q | TRIM2 |
| CMT2S | AD | 11q13.3 | IGHMBP2 |
| CMT2T | AD | 3q25.2 | MME |
| CMT2U | AD | 12q13 | MARS1 |
| CMT2V | AD | 17q11 | NAGLU |
| CMT2W | AD | 5q31 | HARS1 |
| CMT2X | AD | 15q21.1 | SPB11 |
| CMT2Y | AD | 9p13 | VCP |
| CMT2Z | AD | 22q12 | MORC2 |
3.2 Acquired Neuropathies¶
Acquired neuropathies include diabetic neuropathy, amyloidosis, vasculitis, and toxic neuropathies. Diabetic neuropathy is a common cause of acquired neuropathy. Amyloid neuropathy should be considered in cases of small-fiber neuropathy. Vasculitis is a cause of acute and subacute neuropathy. Toxic neuropathies include those caused by medications (e.g., vincristine, cisplatin), alcohol, and heavy metals (e.g., arsenic, lead, thallium). Infections (e.g., HIV, Lyme disease, leprosy) can cause neuropathy. Autoimmune diseases (e.g., Sjögren's syndrome, SLE) can cause neuropathy. Paraneoplastic syndromes can cause neuropathy.
4. CLINICAL FEATURES¶
Patient complaints include numbness, altered sensation to touch (hyperpathia or allodynia), or uncomfortable spontaneous sensations (tingling, burning, or aching). Neuropathic pain can be burning, dull, and poorly localized (protopathic pain), presumably transmitted by polymodal C nociceptor fibers, or sharp and lancinating (epicritic pain), relayed by A-delta fibers. If pain and temperature perception are lost, while vibratory and position sense are preserved along with muscle strength, deep tendon reflexes, and normal nerve conduction studies (NCS), a small-fiber neuropathy is likely. The most likely causes of small-fiber neuropathies, when one is identified, are diabetes mellitus (DM) or glucose intolerance. Amyloid neuropathy should be considered as well in such cases, but most of these small-fiber neuropathies remain idiopathic despite extensive evaluation. Severe proprioceptive loss also narrows the differential diagnosis. Affected patients will note imbalance, especially in the dark. A neurologic examination revealing a dramatic loss of proprioception with vibration loss and normal strength should alert the clinician to consider a sensory neuronopathy/ganglionopathy. In particular, if this loss is asymmetric or affects the arms more than the legs, this pattern suggests a non-length-dependent process as seen in sensory neuronopathies. In patients with generalized symmetric peripheral neuropathy, a standard laboratory evaluation should include a complete blood count, basic chemistries including serum electrolytes and tests of renal and hepatic function, fasting blood glucose (FBS), hemoglobin (Hb) A1c, thyroid function tests, B12, folate, erythrocyte sedimentation rate (ESR), rheumatoid factor, antinuclear antibodies (ANA), serum protein electrophoresis (SPEP) and immunoelectrophoresis or immunofixation, and free light chains in serum and urine. Quantification of the concentration of serum-free light chains and the kappa/lambda ratio is more sensitive than SPEP, immunoelectrophoresis, or immunofixation to detect a monoclonal gammopathy and therefore should be done if amyloidosis is suspected. A skeletal survey should be performed in patients with acquired demyelinating neuropathies and M-spikes to look for osteosclerotic or lytic lesions. Patients with monoclonal gammopathy should also be referred to a hematologist for consideration of a bone marrow biopsy. An oral glucose tolerance test is indicated in patients with painful sensory neuropathies even if FBS and HbA1c are normal, as the test is abnormal in about one-third of such patients.
4.1 History & Physical Examination¶
Seven key questions are asked to help identify the pattern of involvement and the cause of the neuropathy. (1) What systems are involved? (Motor, sensory, autonomic, or combinations). (2) What is the distribution of weakness? (Only distal versus proximal and distal; Focal/asymmetric versus symmetric). (3) What is the nature of the sensory involvement? (Temperature loss or burning or stabbing pain; Vibratory or proprioceptive loss). (4) Is there evidence of upper motor neuron involvement? (Without sensory loss; With sensory loss). (5) What is the temporal evolution? (Acute (days to 4 weeks); Subacute (4–8 weeks); Chronic (>8 weeks); Monophasic, progressive, or relapsing-remitting). (6) Is there evidence for a hereditary neuropathy? (Family history of neuropathy; Lack of sensory symptoms despite sensory signs). (7) Are there any associated medical conditions? (Cancer, diabetes mellitus, connective tissue disease or other autoimmune diseases, infection (e.g., HIV, Lyme disease, leprosy); Medications including over-the-counter drugs that may cause a toxic neuropathy; Preceding events, drugs, toxins).
Table 2 — Table 457-1 Approach to Neuropathic Disorders: Seven Key Questions¶
| Question | Details |
|---|---|
| 1. What systems are involved? | • Motor, sensory, autonomic, or combinations |
| 2. What is the distribution of weakness? | • Only distal versus proximal and distal • Focal/asymmetric versus symmetric |
| 3. What is the nature of the sensory involvement? | • Temperature loss or burning or stabbing pain (e.g., small fiber) • Vibratory or proprioceptive loss (e.g., large fiber) |
| 4. Is there evidence of upper motor neuron involvement? | • Without sensory loss • With sensory loss |
| 5. What is the temporal evolution? | • Acute (days to 4 weeks) • Subacute (4–8 weeks) • Chronic (>8 weeks) • Monophasic, progressive, or relapsing-remitting |
| 6. Is there evidence for a hereditary neuropathy? | • Family history of neuropathy • Lack of sensory symptoms despite sensory signs |
| 7. Are there any associated medical conditions? | • Cancer, diabetes mellitus, connective tissue disease or other autoimmune diseases, infection (e.g., HIV, Lyme disease, leprosy) • Medications including over-the-counter drugs that may cause a toxic neuropathy • Preceding events, drugs, toxins |
4.2 Pattern Recognition¶
Based on the answers to the seven key questions, neuropathic disorders can be classified into several patterns based on the distribution or pattern of sensory, motor, and autonomic involvement. Each pattern has a limited differential diagnosis, and information from laboratory studies usually permits a final diagnosis to be established. Pattern 1: Symmetric proximal and distal weakness with sensory loss. Consider: inflammatory demyelinating polyneuropathy (GBS and CIDP). Pattern 2: Symmetric distal sensory loss with or without distal weakness. Consider: cryptogenic or idiopathic sensory polyneuropathy (CSPN), diabetes mellitus and other metabolic disorders, drugs, toxins, familial (HSAN), CMT, amyloidosis, CANVAS, SORD neuropathy, and others. Pattern 3: Asymmetric distal weakness with sensory loss. With involvement of multiple nerves. Consider: multifocal CIDP, vasculitis, cryoglobulinemia, amyloidosis, sarcoid, infectious (leprosy, Lyme, hepatitis B, C, or E, HIV, CMV), HNPP, tumor infiltration. With involvement of single nerves/regions. Consider: may be any of the above but also could be compressive mononeuropathy, plexopathy, or radiculopathy. Pattern 4: Asymmetric proximal and distal weakness with sensory loss. Consider: polyradiculopathy or plexopathy due to diabetes mellitus, meningeal carcinomatosis or lymphomatosis, sarcoid, amyloid, hereditary plexopathy (HNPP, HNA), idiopathic. Pattern 5: Asymmetric distal weakness without sensory loss. With upper motor neuron findings. Consider: motor neuron disease. Without upper motor neuron findings. Consider: progressive muscular atrophy, juvenile monomelic amyotrophy (Hirayama's disease), multifocal motor neuropathy, multifocal acquired motor axonopathy. Pattern 6: Symmetric sensory loss and distal areflexia with upper motor neuron findings. Consider: vitamin B12, vitamin E, and copper deficiency with combined system degeneration with peripheral neuropathy, chronic liver disease, hereditary leukodystrophies (e.g., adrenomyeloneuropathy), HSP-plus. Pattern 7: Symmetric weakness without sensory loss. With proximal and distal weakness. Consider: SMA. With distal weakness. Consider: hereditary motor neuropathy (distal SMA) or atypical CMT. Pattern 8: Focal midline proximal symmetric weakness. Neck extensor weakness. Consider: ALS. Bulbar weakness. Consider: ALS/PLS, isolated bulbar ALS (IBALS), Kennedy's syndrome (X-linked, bulbospinal SMA), bulbar presentation GBS. Diaphragm weakness (SOB). Consider: ALS. Pattern 9: Asymmetric proprioceptive sensory loss without weakness. Consider causes of a sensory neuronopathy (ganglionopathy): Cancer (paraneoplastic), CANVAS, Sjögren's syndrome, Idiopathic sensory neuronopathy (possible GBS variant), Cisplatin and other chemotherapeutic agents, Vitamin B6 toxicity, HIV-related sensory neuronopathy. Pattern 10: Autonomic symptoms and signs. Consider neuropathies associated with prominent autonomic dysfunction: Hereditary sensory and autonomic neuropathy, Amyloidosis (familial and acquired), Diabetes mellitus, GBS, Idiopathic pandysautonomia (may be a variant of GBS), Porphyria, HIV-related autonomic neuropathy, Vincristine and other chemotherapeutic agents.
Table 3 — Table 457-2 Patterns of Neuropathic Disorders¶
| Pattern | Consider |
|---|---|
| Pattern 1: Symmetric proximal and distal weakness with sensory loss | • Inflammatory demyelinating polyneuropathy (GBS and CIDP) |
| Pattern 2: Symmetric distal sensory loss with or without distal weakness | • Cryptogenic or idiopathic sensory polyneuropathy (CSPN), diabetes mellitus and other metabolic disorders, drugs, toxins, familial (HSAN), CMT, amyloidosis, CANVAS, SORD neuropathy, and others |
| Pattern 3: Asymmetric distal weakness with sensory loss | • With involvement of multiple nerves: Multifocal CIDP, vasculitis, cryoglobulinemia, amyloidosis, sarcoid, infectious (leprosy, Lyme, hepatitis B, C, or E, HIV, CMV), HNPP, tumor infiltration • With involvement of single nerves/regions: May be any of the above but also could be compressive mononeuropathy, plexopathy, or radiculopathy |
| Pattern 4: Asymmetric proximal and distal weakness with sensory loss | • Polyradiculopathy or plexopathy due to diabetes mellitus, meningeal carcinomatosis or lymphomatosis, sarcoid, amyloid, hereditary plexopathy (HNPP, HNA), idiopathic |
| Pattern 5: Asymmetric distal weakness without sensory loss | • With upper motor neuron findings: Motor neuron disease • Without upper motor neuron findings: Progressive muscular atrophy, juvenile monomelic amyotrophy (Hirayama's disease), multifocal motor neuropathy, multifocal acquired motor axonopathy |
| Pattern 6: Symmetric sensory loss and distal areflexia with upper motor neuron findings | • Vitamin B12, vitamin E, and copper deficiency with combined system degeneration with peripheral neuropathy, chronic liver disease, hereditary leukodystrophies (e.g., adrenomyeloneuropathy), HSP-plus |
| Pattern 7: Symmetric weakness without sensory loss | • With proximal and distal weakness: SMA • With distal weakness: Hereditary motor neuropathy (distal SMA) or atypical CMT |
| Pattern 8: Focal midline proximal symmetric weakness | • Neck extensor weakness: ALS • Bulbar weakness: ALS/PLS, isolated bulbar ALS (IBALS), Kennedy's syndrome (X-linked, bulbospinal SMA), bulbar presentation GBS • Diaphragm weakness (SOB): ALS |
| Pattern 9: Asymmetric proprioceptive sensory loss without weakness | • Causes of a sensory neuronopathy (ganglionopathy): Cancer (paraneoplastic), CANVAS, Sjögren's syndrome, Idiopathic sensory neuronopathy (possible GBS variant), Cisplatin and other chemotherapeutic agents, Vitamin B6 toxicity, HIV-related sensory neuronopathy |
| Pattern 10: Autonomic symptoms and signs | • Neuropathies associated with prominent autonomic dysfunction: Hereditary sensory and autonomic neuropathy, Amyloidosis (familial and acquired), Diabetes mellitus, GBS, Idiopathic pandysautonomia (may be a variant of GBS), Porphyria, HIV-related autonomic neuropathy, Vincristine and other chemotherapeutic agents |
5. DIFFERENTIAL DIAGNOSIS¶
The differential diagnosis is based on the pattern of involvement. Pattern 1 (Symmetric proximal and distal weakness with sensory loss) suggests inflammatory demyelinating polyneuropathy (GBS and CIDP). Pattern 2 (Symmetric distal sensory loss with or without distal weakness) suggests cryptogenic or idiopathic sensory polyneuropathy (CSPN), diabetes mellitus and other metabolic disorders, drugs, toxins, familial (HSAN), CMT, amyloidosis, CANVAS, SORD neuropathy, and others. Pattern 3 (Asymmetric distal weakness with sensory loss) suggests multifocal CIDP, vasculitis, cryoglobulinemia, amyloidosis, sarcoid, infectious (leprosy, Lyme, hepatitis B, C, or E, HIV, CMV), HNPP, tumor infiltration, or compressive mononeuropathy, plexopathy, or radiculopathy. Pattern 4 (Asymmetric proximal and distal weakness with sensory loss) suggests polyradiculopathy or plexopathy due to diabetes mellitus, meningeal carcinomatosis or lymphomatosis, sarcoid, amyloid, hereditary plexopathy (HNPP, HNA), idiopathic. Pattern 5 (Asymmetric distal weakness without sensory loss) suggests motor neuron disease (with upper motor neuron findings) or progressive muscular atrophy, juvenile monomelic amyotrophy (Hirayama's disease), multifocal motor neuropathy, multifocal acquired motor axonopathy (without upper motor neuron findings). Pattern 6 (Symmetric sensory loss and distal areflexia with upper motor neuron findings) suggests vitamin B12, vitamin E, and copper deficiency with combined system degeneration with peripheral neuropathy, chronic liver disease, hereditary leukodystrophies (e.g., adrenomyeloneuropathy), HSP-plus. Pattern 7 (Symmetric weakness without sensory loss) suggests SMA (with proximal and distal weakness) or hereditary motor neuropathy (distal SMA) or atypical CMT (with distal weakness). Pattern 8 (Focal midline proximal symmetric weakness) suggests ALS (neck extensor weakness), ALS/PLS, isolated bulbar ALS (IBALS), Kennedy's syndrome, bulbar presentation GBS (bulbar weakness), or ALS (diaphragm weakness). Pattern 9 (Asymmetric proprioceptive sensory loss without weakness) suggests causes of a sensory neuronopathy (ganglionopathy): Cancer (paraneoplastic), CANVAS, Sjögren's syndrome, Idiopathic sensory neuronopathy (possible GBS variant), Cisplatin and other chemotherapeutic agents, Vitamin B6 toxicity, HIV-related sensory neuronopathy. Pattern 10 (Autonomic symptoms and signs) suggests neuropathies associated with prominent autonomic dysfunction: Hereditary sensory and autonomic neuropathy, Amyloidosis (familial and acquired), Diabetes mellitus, GBS, Idiopathic pandysautonomia (may be a variant of GBS), Porphyria, HIV-related autonomic neuropathy, Vincristine and other chemotherapeutic agents.
6. INVESTIGATIONS & DIAGNOSIS¶
The electrodiagnostic (EDx) evaluation of patients with a suspected peripheral neuropathy consists of NCS and needle electromyography (EMG). In addition, studies of autonomic function can be valuable. The electrophysiologic data can confirm whether the neuropathic disorder is a mononeuropathy, multiple mononeuropathy (mononeuropathy multiplex), radiculopathy, plexopathy, or generalized polyneuropathy. Similarly, EDx evaluation can ascertain whether the process involves only sensory fibers, motor fibers, autonomic fibers, or a combination of these. Finally, the electrophysiologic data can help distinguish axonopathies from myelinopathies as well as axonal degeneration secondary to ganglionopathies from the more common length-dependent axonopathies. NCS are most helpful in classifying a neuropathy as due to axonal degeneration or segmental demyelination. In general, low-amplitude potentials with relatively preserved distal latencies, conduction velocities, and late potentials, along with fibrillations on needle EMG, suggest an axonal neuropathy. On the other hand, slow conduction velocities, prolonged distal latencies and late potentials, relatively preserved amplitudes, and the absence of fibrillations on needle EMG imply a primary demyelinating neuropathy. The presence of nonuniform slowing of conduction velocity, conduction block, or temporal dispersion further suggests an acquired demyelinating neuropathy (e.g., GBS or CIDP) as opposed to a hereditary demyelinating neuropathy (e.g., CMT type 1). Autonomic studies are used to assess small myelinated (A-delta) or unmyelinated (C) nerve fiber involvement. Such testing includes heart rate response to deep breathing, heart rate and blood pressure response to both the Valsalva maneuver and tilt-table testing, and quantitative sudomotor axon reflex testing. These studies are particularly useful in patients who have pure small-fiber neuropathy or autonomic neuropathy in which routine NCS are normal. There are many autoantibody panels (various antiganglioside antibodies) marketed for screening routine neuropathy patients for a treatable condition. These autoantibodies have no proven clinical utility or added benefit beyond the information obtained from a complete clinical examination and detailed EDx. A heavy metal screen is also not necessary as a screening procedure, unless there is a history of possible exposure or suggestive features on examination (e.g., severe painful sensorimotor and autonomic neuropathy and alopecia—thallium; severe painful sensorimotor neuropathy with or without gastrointestinal [GI] disturbance and Mee's lines—arsenic; wrist or finger extensor weakness and anemia with basophilic stippling of red blood cells—lead). In patients with suspected GBS or CIDP, a lumbar puncture is indicated to look for an elevated cerebrospinal fluid (CSF) protein. In idiopathic cases of GBS and CIDP, CSF pleocytosis is usually absent. If cells are present, one should consider HIV infection, Lyme disease, sarcoidosis, or lymphomatous or leukemic infiltration of nerve roots. Recently, serum IgG4 antibodies to neurofascin and contactin-2 have been discovered in CIDP with severe sensory ataxia, tremor, and distal weakness. These cases are difficult to treat with standard immunotherapies but may respond to rituximab. Some patients with GBS and CIDP have abnormal liver function tests. In these cases, it is important to also check for hepatitis B and C, HIV, CMV, and Epstein-Barr virus (EBV) infection. In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren's syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren's syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without other manifestations of Sjögren's syndrome. Thus, patients with sensory ataxia should be tested for antibodies to SS-A/Ro and SS-B/La, in addition to the routine ANA. To evaluate a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained. These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are also present with breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of cancer, and detection of these autoantibodies should lead to a search for malignancy. In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren's syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren's syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without other manifestations of Sjögren's syndrome. Thus, patients with sensory ataxia should be tested for antibodies to SS-A/Ro and SS-B/La, in addition to the routine ANA. To evaluate a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained. These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are also present with breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of cancer, and detection of these autoantibodies should lead to a search for malignancy.
6.1 Electrodiagnostic Studies¶
The electrodiagnostic (EDx) evaluation of patients with a suspected peripheral neuropathy consists of NCS and needle electromyography (EMG). In addition, studies of autonomic function can be valuable. The electrophysiologic data can confirm whether the neuropathic disorder is a mononeuropathy, multiple mononeuropathy (mononeuropathy multiplex), radiculopathy, plexopathy, or generalized polyneuropathy. Similarly, EDx evaluation can ascertain whether the process involves only sensory fibers, motor fibers, autonomic fibers, or a combination of these. Finally, the electrophysiologic data can help distinguish axonopathies from myelinopathies as well as axonal degeneration secondary to ganglionopathies from the more common length-dependent axonopathies. NCS are most helpful in classifying a neuropathy as due to axonal degeneration or segmental demyelination. In general, low-amplitude potentials with relatively preserved distal latencies, conduction velocities, and late potentials, along with fibrillations on needle EMG, suggest an axonal neuropathy. On the other hand, slow conduction velocities, prolonged distal latencies and late potentials, relatively preserved amplitudes, and the absence of fibrillations on needle EMG imply a primary demyelinating neuropathy. The presence of nonuniform slowing of conduction velocity, conduction block, or temporal dispersion further suggests an acquired demyelinating neuropathy (e.g., GBS or CIDP) as opposed to a hereditary demyelinating neuropathy (e.g., CMT type 1). Autonomic studies are used to assess small myelinated (A-delta) or unmyelinated (C) nerve fiber involvement. Such testing includes heart rate response to deep breathing, heart rate and blood pressure response to both the Valsalva maneuver and tilt-table testing, and quantitative sudomotor axon reflex testing. These studies are particularly useful in patients who have pure small-fiber neuropathy or autonomic neuropathy in which routine NCS are normal.
Table 4 — Table 457-3 Electrophysiologic Features: Axonal Degeneration versus Segmental Demyelination¶
| Parameter | Axonal Degeneration | Segmental Demyelination |
|---|---|---|
| Motor Nerve Conduction Studies | ||
| CMAP amplitude | Decreased | Normal (except with CB or distal dispersion) |
| Distal latency | Normal | Prolonged |
| Conduction velocity | Normal | Slow |
| Conduction block | Absent | Present |
| Temporal dispersion | Absent | Present |
| F wave | Normal or absent | Prolonged or absent |
| H reflex | Normal or absent | Prolonged or absent |
| Sensory Nerve Conduction Studies | ||
| SNAP amplitude | Decreased | Normal or decreased |
| Distal latency | Normal | Prolonged |
| Conduction velocity | Normal | Slow |
| Needle EMG | ||
| Spontaneous activity | ||
| Fibrillations | Present | Absent |
| Fasciculations | Present | Absent |
| Motor unit potentials | ||
| Recruitment | Decreased | Decreased |
| Morphology | Long duration, large amplitude, polyphasic (if there is reinnervation) | Normal |
6.2 Other Laboratory & Imaging¶
In patients with generalized symmetric peripheral neuropathy, a standard laboratory evaluation should include a complete blood count, basic chemistries including serum electrolytes and tests of renal and hepatic function, fasting blood glucose (FBS), hemoglobin (Hb) A1c, thyroid function tests, B12, folate, erythrocyte sedimentation rate (ESR), rheumatoid factor, antinuclear antibodies (ANA), serum protein electrophoresis (SPEP) and immunoelectrophoresis or immunofixation, and free light chains in serum and urine. Quantification of the concentration of serum-free light chains and the kappa/lambda ratio is more sensitive than SPEP, immunoelectrophoresis, or immunofixation to detect a monoclonal gammopathy and therefore should be done if amyloidosis is suspected. A skeletal survey should be performed in patients with acquired demyelinating neuropathies and M-spikes to look for osteosclerotic or lytic lesions. Patients with monoclonal gammopathy should also be referred to a hematologist for consideration of a bone marrow biopsy. An oral glucose tolerance test is indicated in patients with painful sensory neuropathies even if FBS and HbA1c are normal, as the test is abnormal in about one-third of such patients. In addition to the above tests, patients with a mononeuropathy multiplex pattern of involvement should have a vasculitis workup, including immunologic staining can be used to measure the density of small unmyelinated fibers. The density of these nerve fibers is reduced in patients with small-fiber neuropathies in whom NCS and routine nerve biopsies are often normal. This technique may allow for an objective measurement in patients with mainly subjective symptoms. However, it often adds little to what one already knows from the clinical examination and EDx. There are many autoantibody panels (various antiganglioside antibodies) marketed for screening routine neuropathy patients for a treatable condition. These autoantibodies have no proven clinical utility or added benefit beyond the information obtained from a complete clinical examination and detailed EDx. A heavy metal screen is also not necessary as a screening procedure, unless there is a history of possible exposure or suggestive features on examination (e.g., severe painful sensorimotor and autonomic neuropathy and alopecia—thallium; severe painful sensorimotor neuropathy with or without gastrointestinal [GI] disturbance and Mee's lines—arsenic; wrist or finger extensor weakness and anemia with basophilic stippling of red blood cells—lead). In patients with suspected GBS or CIDP, a lumbar puncture is indicated to look for an elevated cerebrospinal fluid (CSF) protein. In idiopathic cases of GBS and CIDP, CSF pleocytosis is usually absent. If cells are present, one should consider HIV infection, Lyme disease, sarcoidosis, or lymphomatous or leukemic infiltration of nerve roots. Recently, serum IgG4 antibodies to neurofascin and contactin-2 have been discovered in CIDP with severe sensory ataxia, tremor, and distal weakness. These cases are difficult to treat with standard immunotherapies but may respond to rituximab. Some patients with GBS and CIDP have abnormal liver function tests. In these cases, it is important to also check for hepatitis B and C, HIV, CMV, and Epstein-Barr virus (EBV) infection. In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren's syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren's syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without other manifestations of Sjögren's syndrome. Thus, patients with sensory ataxia should be tested for antibodies to SS-A/Ro and SS-B/La, in addition to the routine ANA. To evaluate a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained. These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are also present with breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of cancer, and detection of these autoantibodies should lead to a search for malignancy.
6.3 Biopsy¶
Nerve biopsies are now rarely performed in the evaluation of neuropathies. The primary indication for nerve biopsy is suspicion for amyloid neuropathy or vasculitis. In most instances, the abnormalities present on biopsies do not help distinguish one form of peripheral neuropathy from another (beyond what is already apparent by clinical examination and the NCS). Nerve biopsies should only be performed when the NCS are abnormal. The sural nerve is most commonly biopsied because it is a pure sensory nerve and biopsy will not result in loss of motor function. In suspected vasculitis, a combination biopsy of a superficial peroneal nerve (pure sensory) and the underlying peroneus brevis muscle obtained from a single small incision increases the diagnostic yield. Tissue can be analyzed to assess for evidence of inflammation, vasculitis, or amyloid deposition. Semithin plastic sections, teased fiber preparations, and electron microscopy are used to assess the morphology of the nerve fibers and to distinguish axonopathies from myelinopathies. Skin biopsies are sometimes used to diagnose a small-fiber neuropathy. Following a punch biopsy of the skin in the distal lower extremity, immunologic staining can be used to measure the density of small unmyelinated fibers. The density of these nerve fibers is reduced in patients with small-fiber neuropathies in whom NCS and routine nerve biopsies are often normal. This technique may allow for an objective measurement in patients with mainly subjective symptoms. However, it often adds little to what one already knows from the clinical examination and EDx.
7. MANAGEMENT & TREATMENT¶
There are no medical therapies for any of the CMTs, but physical and occupational therapy can be beneficial, as can bracing (e.g., ankle-foot orthotics for foot drop) and other orthotic devices. In patients with suspected GBS or CIDP, treatment includes IVIg or plasmapheresis. In patients with suspected GBS or CIDP, a lumbar puncture is indicated to look for an elevated cerebrospinal fluid (CSF) protein. In idiopathic cases of GBS and CIDP, CSF pleocytosis is usually absent. If cells are present, one should consider HIV infection, Lyme disease, sarcoidosis, or lymphomatous or leukemic infiltration of nerve roots. Recently, serum IgG4 antibodies to neurofascin and contactin-2 have been discovered in CIDP with severe sensory ataxia, tremor, and distal weakness. These cases are difficult to treat with standard immunotherapies but may respond to rituximab. Some patients with GBS and CIDP have abnormal liver function tests. In these cases, it is important to also check for hepatitis B and C, HIV, CMV, and Epstein-Barr virus (EBV) infection. In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren's syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren's syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without other manifestations of Sjögren's syndrome. Thus, patients with sensory ataxia should be tested for antibodies to SS-A/Ro and SS-B/La, in addition to the routine ANA. To evaluate a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained. These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are also present with breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of cancer, and detection of these autoantibodies should lead to a search for malignancy. In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren's syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren's syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without other manifestations of Sjögren's syndrome. Thus, patients with sensory ataxia should be tested for antibodies to SS-A/Ro and SS-B/La, in addition to the routine ANA. To evaluate a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained. These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are also present with breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of cancer, and detection of these autoantibodies should lead to a search for malignancy.
7.1 Hereditary Neuropathies¶
There are no medical therapies for any of the CMTs, but physical and occupational therapy can be beneficial, as can bracing (e.g., ankle-foot orthotics for foot drop) and other orthotic devices. CMT1 is the most common form of hereditary neuropathy. Affected individuals usually present in the first to third decade of life with distal leg weakness (e.g., foot drop), although patients may remain asymptomatic even late in life. People with CMT generally do not complain of numbness or tingling, which can be helpful in distinguishing CMT from acquired forms of neuropathy in which sensory symptoms usually predominate. Although usually asymptomatic, reduced sensation to all modalities is apparent on examination. Muscle stretch reflexes are unobtainable or reduced throughout. There is often atrophy of the muscles below the knee (particularly the anterior compartment), leading to so-called inverted champagne bottle legs. Motor NCVs are generally in the 20–25 m/s range. Nerve biopsies usually are not performed on patients suspected of having CMT1, because the diagnosis usually can be made by less invasive testing (e.g., NCS and genetic studies). However, when done, the biopsies reveal the reduced numbers of myelinated nerve fibers with a predilection for loss of large-diameter fibers and Schwann cell proliferation around thinly myelinated or demyelinated fibers, forming so-called onion bulbs. CMT1A is the most common subtype of CMT1, representing 70% of cases, and is caused by a 1.5-Mb duplication within chromosome 17p11.2-12 encoding the gene for peripheral myelin protein-22 (PMP-22). This results in patients having three copies of the PMP-22 gene rather than two. This protein accounts for 2–5% of myelin protein and is expressed in compact regions of the peripheral myelin sheath. Approximately 20% of patients with CMT1 have CMT1B, caused by mutations in the myelin protein zero (MPZ). CMT1B is for the most part clinically, electrophysiologically, and histologically indistinguishable from CMT1A. MPZ is an integral myelin protein and accounts for more than half of the myelin protein in peripheral nerves. Other forms of CMT1 are much less common and also indistinguishable from one another clinically and electrophysiologically. CMT2 occurs approximately half as frequently as CMT1, and CMT2 tends to present later in life. Affected individuals usually become symptomatic in the second decade; some cases present earlier in childhood, whereas others remain asymptomatic into late adult life. Clinically, CMT2 is for the most part indistinguishable from CMT1. NCS are helpful in this regard; in contrast to CMT1, the velocities are normal or near normal.
7.2 Acquired Neuropathies¶
In patients with suspected GBS or CIDP, treatment includes IVIg or plasmapheresis. In patients with suspected GBS or CIDP, a lumbar puncture is indicated to look for an elevated cerebrospinal fluid (CSF) protein. In idiopathic cases of GBS and CIDP, CSF pleocytosis is usually absent. If cells are present, one should consider HIV infection, Lyme disease, sarcoidosis, or lymphomatous or leukemic infiltration of nerve roots. Recently, serum IgG4 antibodies to neurofascin and contactin-2 have been discovered in CIDP with severe sensory ataxia, tremor, and distal weakness. These cases are difficult to treat with standard immunotherapies but may respond to rituximab. Some patients with GBS and CIDP have abnormal liver function tests. In these cases, it is important to also check for hepatitis B and C, HIV, CMV, and Epstein-Barr virus (EBV) infection. In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren's syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren's syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without other manifestations of Sjögren's syndrome. Thus, patients with sensory ataxia should be tested for antibodies to SS-A/Ro and SS-B/La, in addition to the routine ANA. To evaluate a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained. These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are also present with breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of cancer, and detection of these autoantibodies should lead to a search for malignancy.
8. PROGNOSIS & COMPLICATIONS¶
CMT disease is a syndrome of many genetically distinct disorders. The various subtypes of CMT are classified according to the nerve conduction velocities (NCVs) and predominant pathology (e.g., demyelination or axonal degeneration), inheritance pattern (autosomal dominant, autosomal recessive, or X-linked), and the specific mutated genes. Type 1 CMT (or CMT1) refers to inherited demyelinating sensorimotor neuropathies, whereas the axonal sensory neuropathies are classified as CMT2. By definition, motor conduction velocities in the arms are slowed to 38 m/s in CMT2. However, most cases of CMT1 actually have motor NCVs between 20 and 25 m/s. CMT1 and CMT2 usually begin in childhood or early adult life; however, onset later in life can occur, particularly in CMT2. Both are inherited in an autosomal dominant fashion, with a few exceptions. There are no medical therapies for any of the CMTs, but physical and occupational therapy can be beneficial, as can bracing (e.g., ankle-foot orthotics for foot drop) and other orthotic devices. In patients with suspected GBS or CIDP, treatment includes IVIg or plasmapheresis. In patients with suspected GBS or CIDP, a lumbar puncture is indicated to look for an elevated cerebrospinal fluid (CSF) protein. In idiopathic cases of GBS and CIDP, CSF pleocytosis is usually absent. If cells are present, one should consider HIV infection, Lyme disease, sarcoidosis, or lymphomatous or leukemic infiltration of nerve roots. Recently, serum IgG4 antibodies to neurofascin and contactin-2 have been discovered in CIDP with severe sensory ataxia, tremor, and distal weakness. These cases are difficult to treat with standard immunotherapies but may respond to rituximab. Some patients with GBS and CIDP have abnormal liver function tests. In these cases, it is important to also check for hepatitis B and C, HIV, CMV, and Epstein-Barr virus (EBV) infection. In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren's syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren's syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without other manifestations of Sjögren's syndrome. Thus, patients with sensory ataxia should be tested for antibodies to SS-A/Ro and SS-B/La, in addition to the routine ANA. To evaluate a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained. These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are also present with breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of cancer, and detection of these autoantibodies should lead to a search for malignancy.
9. SPECIAL CONSIDERATIONS¶
In patients with suspected GBS or CIDP, treatment includes IVIg or plasmapheresis. In patients with suspected GBS or CIDP, a lumbar puncture is indicated to look for an elevated cerebrospinal fluid (CSF) protein. In idiopathic cases of GBS and CIDP, CSF pleocytosis is usually absent. If cells are present, one should consider HIV infection, Lyme disease, sarcoidosis, or lymphomatous or leukemic infiltration of nerve roots. Recently, serum IgG4 antibodies to neurofascin and contactin-2 have been discovered in CIDP with severe sensory ataxia, tremor, and distal weakness. These cases are difficult to treat with standard immunotherapies but may respond to rituximab. Some patients with GBS and CIDP have abnormal liver function tests. In these cases, it is important to also check for hepatitis B and C, HIV, CMV, and Epstein-Barr virus (EBV) infection. In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren's syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren's syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without other manifestations of Sjögren's syndrome. Thus, patients with sensory ataxia should be tested for antibodies to SS-A/Ro and SS-B/La, in addition to the routine ANA. To evaluate a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained. These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are also present with breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of cancer, and detection of these autoantibodies should lead to a search for malignancy.
10. KEY PEARLS & CLINICAL TRAPS¶
Approximately half of patients with generalized symmetric peripheral neuropathy have no identifiable etiology. CMT1 is the most common form of hereditary neuropathy. Small-fiber neuropathy is likely if pain and temperature perception are lost while vibratory and position sense are preserved, with normal NCS. Vitamin B12 deficiency is the most common cause of combined system degeneration with neuropathy and upper motor neuron involvement. Nerve biopsy is rarely performed now; primary indications are suspicion for amyloid neuropathy or vasculitis. Skin biopsy can diagnose small-fiber neuropathy by measuring density of small unmyelinated fibers. In patients with suspected GBS or CIDP, CSF pleocytosis is usually absent; if cells are present, consider HIV, Lyme, sarcoidosis, or lymphomatous infiltration. CMT1A is caused by a 1.5-Mb duplication within chromosome 17p11.2-12 encoding the gene for peripheral myelin protein-22 (PMP-22). Autonomic dysfunction in the absence of diabetes should alert the clinician to the possibility of amyloid polyneuropathy. Severe proprioceptive loss also narrows the differential diagnosis. Affected patients will note imbalance, especially in the dark. A neurologic examination revealing a dramatic loss of proprioception with vibration loss and normal strength should alert the clinician to consider a sensory neuronopathy/ganglionopathy. In particular, if this loss is asymmetric or affects the arms more than the legs, this pattern suggests a non-length-dependent process as seen in sensory neuronopathies. In patients with generalized symmetric peripheral neuropathy, a standard laboratory evaluation should include a complete blood count, basic chemistries including serum electrolytes and tests of renal and hepatic function, fasting blood glucose (FBS), hemoglobin (Hb) A1c, thyroid function tests, B12, folate, erythrocyte sedimentation rate (ESR), rheumatoid factor, antinuclear antibodies (ANA), serum protein electrophoresis (SPEP) and immunoelectrophoresis or immunofixation, and free light chains in serum and urine. Quantification of the concentration of serum-free light chains and the kappa/lambda ratio is more sensitive than SPEP, immunoelectrophoresis, or immunofixation to detect a monoclonal gammopathy and therefore should be done if amyloidosis is suspected. A skeletal survey should be performed in patients with acquired demyelinating neuropathies and M-spikes to look for osteosclerotic or lytic lesions. Patients with monoclonal gammopathy should also be referred to a hematologist for consideration of a bone marrow biopsy. An oral glucose tolerance test is indicated in patients with painful sensory neuropathies even if FBS and HbA1c are normal, as the test is abnormal in about one-third of such patients.
Flowcharts & Algorithms¶
Reproduced from Harrison's 22nd Edition.
Flowchart 1¶
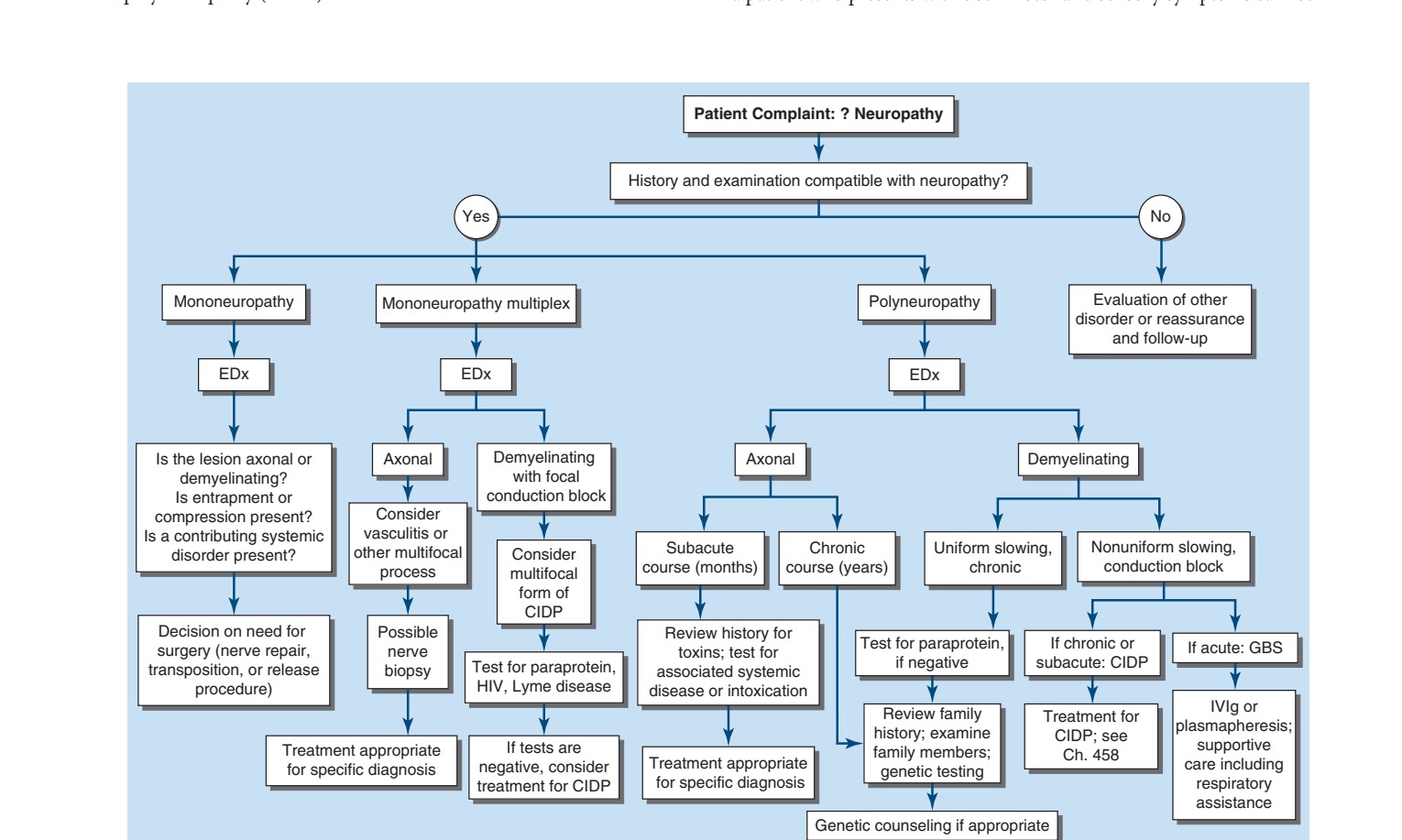
Caption: FIGURE 457-1 Approach to the evaluation of peripheral neuropathies. CIDP, chronic Guillain-Barré syndrome; IVIg, intravenous immunoglobulin.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶
Caption: FIGURE 457-3 Lumbosacral plexus. (Reproduced with permission from AA Amato, JA Russell (eds): Neuromuscular Disorders, 2nd ed. New York: McGraw-Hill Education; 2016.) — Figure 457-1: Algorithmic approach to the evaluation of peripheral neuropathies, illustrating the seven key questions to identify lesion location, cause, and treatment needs, with decision nodes for mononeuropathy, polyneuropathy, axonal vs demyelinating, and specific etiologies like GBS/CIDP.
Figure 2¶

Caption: FIGURE 457-2 Brachial plexus anatomy. L, lateral; M, medial; P, posterior. (Reproduced Baltimore, Williams and Wilkins, 1974.) — Figure 457-2: Classification of Charcot-Marie-Tooth disease and related neuropathies, displaying inheritance patterns (autosomal dominant, recessive, X-linked), gene locations, and specific gene mutations for CMT1, CMT2, and CMT4 subtypes.
Figure 3¶

Caption: FIGURE 457-3 Lumbosacral plexus. (Reproduced with permission from AA Amato, JA Russell (eds): Neuromuscular Disorders, 2nd ed. New York: McGraw-Hill Education; 2016.) — Figure 457-3: Electrophysiologic features distinguishing axonal degeneration from segmental demyelination, showing motor and sensory nerve conduction study parameters (CMAP amplitude, distal latency, conduction velocity) and needle EMG findings (fibrillations, motor unit potentials).
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.