Cancer Genetics¶
Chapter 76 | Harrison's 22e · Part 4 – Oncology: Solid Tumors
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- See source text for full details
📑 Table of Contents¶
📋 Figures in This Chapter¶
RAW CONTENT¶
[PAGE 519] Cancer Genetics 519 CHAPTER 76 Manson JE et al: Vitamin D supplements and prevention of cancer and THE CLONAL ORIGIN AND MULTISTEP cardiovascular disease. N Engl J Med 380:33, 2019. NATURE OF CANCER Mcneil JJ et al: Effect of aspirin on all-cause mortality in the healthy Nearly all cancers originate from a single cell; this clonal origin is a elderly. N Engl J Med 379:1519, 2018. critical discriminating feature between neoplasia and hyperplasia. Melnikow J et al: Screening for cervical cancer with high-risk human Multiple cumulative mutational events are invariably required for the papillomavirus testing: Updated evidence report and systematic progression of a tumor from normal to fully malignant phenotype. The review for the US Preventive Services Task Force. JAMA 320:687, process can be seen as Darwinian microevolution in which, at each 2018. successive step, the mutated cells gain a growth advantage resulting Welch HG et al: Epidemiologic signatures in cancer. N Engl J Med in the expansion of a neoplastic clone (Fig. 76-1). Based on observa- 384:14, 2019. tions of cancer frequency increases during aging, the epidemiologists Woloshin S et al: Breast-cancer mortality trends in four countries Armitage and Doll and Nordling independently proposed that cancer with varied screening practices. N Engl J Med 389:1061, 2023. is a result of three discrete cellular changes. Remarkably, this early Zeraatkar D et al: Effect of lower versus high red meat intake on model has been validated by extensive sequencing of cancer genomes. cardiometabolic and cancer outcomes: A systematic review of ran- These studies revealed that just three causal mutations are required for domized trials. Ann Intern Med 171:721, 2019. the development of several of the most common cancers. Overall, it is currently believed that most common solid tumors require a minimum of three mutated cancer driver genes (either oncogenes or tumor- suppressor genes) for their development. One or two mutations are sufficient for benign tumorigenesis, but not for the invasive capacity that distinguishes cancers from benign tumors. Less common tumors, such as liquid tumors (leukemias or lymphomas), sarcomas, and 76 Cancer Genetics childhood tumors, appear to require only two driver gene alterations for malignancy. Note that a cancer driver gene is best defined as one containing a mutation that increases the selective growth advantage of Fred Bunz, Bert Vogelstein the cell containing it. Normally, cell birth and cell death are in perfect equilibrium; every time a cell is born, another in the same lineage dies. Cancer driver gene mutations alter this equilibrium, so that more cells CANCER IS A GENETIC DISEASE are born than die. The imbalance is often slight, so that the difference Cancer arises through a series of somatic alterations in DNA that result between cell birth and cell death can be less than 1%. This explains, in in unrestrained cellular proliferation. Most of these alterations involve combination with the low rate of mutation, why tumorigenesis—the subtle sequence changes in DNA (i.e., mutations). The somatic mutations journey from a normal cell to a typical malignant, solid tumor—often may originate as a consequence of random replication errors or exposure takes decades. to carcinogens (e.g., radiation) and can be exacerbated by faulty DNA We now know the precise nature of the genetic alterations respon- repair processes. While most cancers arise sporadically, clustering of can- sible for nearly all malignancies and are beginning to understand how cers occurs in families that carry a germline mutation in a cancer gene. these alterations promote the distinct stages of tumor growth. The prototypical example is colon cancer, in which analyses of genomes HISTORICAL PERSPECTIVE from the entire spectrum of neoplastic growths—from normal colon The idea that cancer progression is driven by sequential somatic muta- tions in specific genes has only gained general acceptance in the past 30 years. Before the advent of the microscope, cancer was believed to be composed of aggregates of mucus or other noncellular matter. By the middle of the nineteenth century, it became clear that tumors were masses of cells and that these cells arose from the normal cells of the Initiation tissue from which the cancer originated. The molecular basis for the uncontrolled proliferation of cancer cells was to remain a mystery for another century. During that time, a number of theories for the origin of cancer were postulated. The great biochemist Otto Warburg proposed the combustion theory of cancer, which stipulated that cancer was due to abnormal oxygen metabolism. Others believed that all cancers were caused by viruses and that cancer was in fact a contagious disease. In the end, observations of cancer occurring in chimney sweeps, Expansion studies of x-rays, and the overwhelming data demonstrating cigarette smoke as a causative agent in lung cancer, together with Ames’s work on chemical mutagenesis, were consistent with the idea that cancer originated through changes in DNA. However, it was not until the somatic mutations responsible for cancer were identified at the molec- ular level that the genetic basis of cancer was definitively established. Although the viral theory of cancer did not prove to be generally Invasion accurate (with exceptions such as human papillomaviruses, which can cause cervical and other cancers), the study of retroviruses led to the discovery of the first human oncogenes in the late 1970s. Oncogenes are one of the two major classes of cancer driver genes. The study of families with genetic predisposition to cancer was instrumental to the discovery of the other major class of cancer driver genes, called tumor- FIGURE 76-1 Multistep clonal development of malignancy. In this diagram, a series of three cumulative mutations, each with a modest growth advantage acting alone, suppressor genes. Current technologies permit the sequence analysis of eventually results in a malignant tumor. Note that not all such alterations result in entire cancer genomes and provide a comprehensive view of the genetic progression. The actual number of cumulative mutations necessary to transform changes that cause tumors to arise and become malignant. The field from the normal to the malignant state has been estimated to be three for several of that studies the various types of mutations, as well as the consequences the most common types of cancer. (Adapted and modified from PC Nowell: The clonal of these mutations in tumor cells, is now known as cancer genetics. evolution of tumor cell populations. Science 194:23, 1976.)
[TABLE] Initiation Expansion Invasion [/TABLE]
[PAGE 520] 520 PART 4 Oncology and Hematology promote tumor formation. The agent responsible Microsatellite Instability (MIN) or Chromosomal Instability (CIN) for the transmission of the cancer was a retrovirus (Rous sarcoma virus [RSV]), and the oncogene responsible was identified 75 years later as V-SRC. Other oncogenes were also discovered through APC inactivation KRAS or SMAD4 or TGFb II their presence in the genomes of retroviruses that or b-catenin BRAF inactivation are capable of causing cancers in chickens, mice, activation activation TP53 inactivation and rats. The nonmutated cellular homologues of these viral genes are called proto-oncogenes and are often targets of mutation or aberrant regula- tion in human cancer. Whereas many oncogenes were discovered on the basis of their presence in Normal Early Late Carcinoma Metastasis retroviruses, other oncogenes, particularly those epithelium adenoma adenoma involved in translocations characteristic of par- ticular leukemias and lymphomas, were identi- Initiation Expansion Invasion fied through genomic approaches. Investigators FIGURE 76-2 Progressive somatic mutational steps in the development of colon carcinoma. The cloned the sequences surrounding the chromo- accumulation of alterations
Flowcharts & Algorithms¶
Reproduced from Harrison's 22nd Edition.
Flowchart 1¶

Caption: FIGURE 76-6 Algorithm for genetic testing in a family with cancer predisposition. The key step is the identification of a disease mutation in a cancer patient, which in is an indication for the testing of asymptomatic family members. Asymptomatic as family members who test positive may require increased screening or surgery, whereas those who test negative are at no greater risk for cancer than the general population. It should be emphasized that no molecular assay used for this sort of testing is 100% sensitive; negative results must be interpreted with this caveat in mind.
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 76-1 Multistep clonal development of malignancy. In this diagram, a series of three cumulative mutations, each with a modest growth advantage acting alone, of eventually results in a malignant tumor. Note that not all such alterations result in progression. The actual number of cumulative mutations necessary to transform from the normal to the malignant state has been estimated to be three for several of the most common types of cancer. (Adapted and modified from PC Nowell: The clonal evolution of tumor cell populations. Science 194:23, 1976.)
Figure 2¶

Caption: FIGURE 76-5 Germline and somatic mutations in the tumor-suppressor gene domains: an oligomerization region (O), armadillo repeats (ARM), 15-amino-acid repeats binding EB1 and the Drosophila discs large homologue (E/D). Shown are 650 somatic and gene (from the APC database at www.umd.be/APC). All known pathogenic mutations of be relatively evenly distributed up to codon 1600 except for two mutation hotspots mutations found in familial adenomatous polyposis (FAP) families.
Figure 3¶
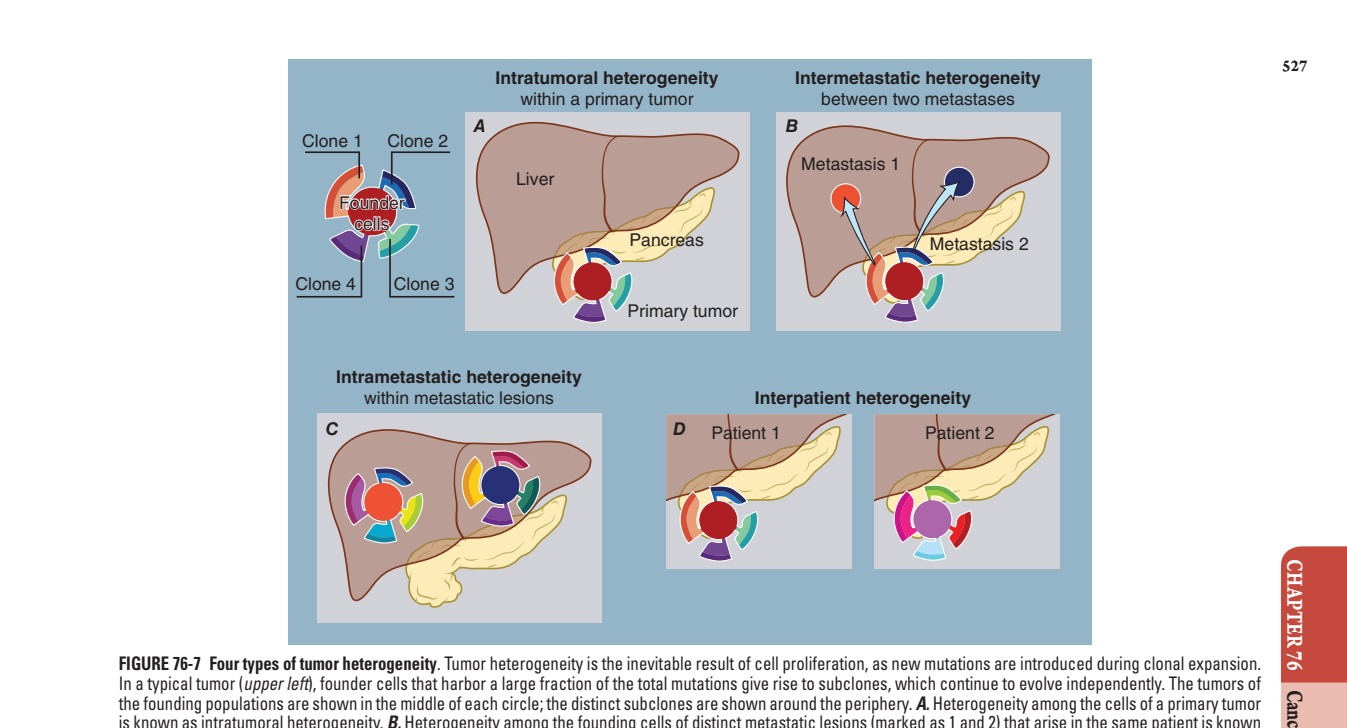
Caption: FIGURE 76-7 Four types of tumor heterogeneity. Tumor heterogeneity is the inevitable In a typical tumor (upper left), founder cells that harbor a large fraction of the total the founding populations are shown in the middle of each circle; the distinct subclones are is known as intratumoral heterogeneity. B. Heterogeneity among the founding cells of as intermetastatic heterogeneity. C. Heterogeneity among the cells of each metastatic The mutations in the tumors of two patients are almost completely distinct. 339(6127):1546, 2013.)
Figure 4¶

Caption: CHAPTER 76 22. Figure 76-3 illustrates the genera- its protein product. The consequence of gene product is the activation of signal to cell growth independent of normal drug that specifically blocks the activity remarkable efficacy with little tox- successful targeting of BCR-ABL by targeted anticancer therapies. IN SOLID
Figure 5¶

Caption: FIGURE 76-2 Progressive somatic mutational steps in the development of colon accumulation of alterations in a number of different genes results in the progression from through adenoma to full-blown carcinoma. Genetic instability (microsatellite or the progression by increasing the likelihood of mutation at each step. Patients with are already one step into this pathway because they inherit a germline alteration of transforming growth factor. epithelium through adenoma to carcinoma—have identified mutations that are highly characteristic of each type of lesion (Fig. 76-2).
Figure 6¶

Caption: FIGURE 76-3 Specific translocation seen in chronic myeloid leukemia (CML). The chromosomes 9 and 22 with the breakpoint joining the sequences of the ABL oncogene entirely novel fusion protein with modified function.
Figure 7¶
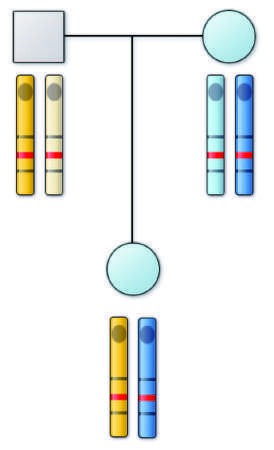
Caption: FIGURE 76-4 Diagram of possible mechanisms for tumor formation in an individual with hereditary (familial) retinoblastoma. On the left is shown the affected individual who has inherited the abnormal (Rb) allele from her affected mother. The normal allele is shown as a (+). The four chromosomes of her two drawn to indicate their origin. Flanking the retinoblastoma locus are genetic markers (A and B) also analyzed in this family. Markers A3 and B3 are on the carrying the retinoblastoma disease gene. Tumor formation results when the normal allele, which this patient inherited from her father, is inactivated. On the four possible ways in which this could occur. In each case, the resulting chromosome 13 arrangement is shown. Note that in the first three situations, the has been lost in the tumor tissue, which is referred to as loss of heterozygosity (LOH) at this locus.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.