Pituitary Tumor Syndromes¶
Chapter 392 | Part 12: Endocrinology and Metabolism · Part 12 – Endocrinology & Metabolism
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- Pituitary adenomas are the most common cause of pituitary hormone hypersecretion and hyposecretion syndromes in adults, accounting for ~15% of all intracranial neoplasms.
- Hyperprolactinemia is the most common pituitary hormone hypersecretion syndrome; PRL levels >200 μg/L suggest a prolactinoma, while levels <200 μg/L are often due to stalk compression or drugs.
- Visual field defects (bitemporal hemianopia) are the hallmark of suprasellar extension compressing the optic chiasm; nasal fibers are most vulnerable.
- MRI with gadolinium is the imaging modality of choice; adenomas typically show lower density than normal tissue on T1 and increased signal on T2.
- Transsphenoidal surgery is the preferred surgical approach for most pituitary tumors; radiation therapy is reserved for residual tumor or nonfunctioning tumors.
- MEN1 syndrome (MENIN mutation) is associated with pituitary adenomas in ~40% of patients, often prolactinomas.
- Familial acromegaly is associated with germline mutations in the AIP gene (aryl hydrocarbon receptor interacting protein).
- Craniopharyngiomas are benign, suprasellar cystic masses derived from Rathke's pouch; ~75% recur without radiotherapy.
- Drug-induced hyperprolactinemia is common with antipsychotics (e.g., risperidone), methyldopa, and verapamil.
- GnRH testing (100 μg IV) distinguishes hypothalamic from pituitary causes of hypogonadism; a normal response suggests intact pituitary function.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 Sellar Mass Classification
- 1.2 Pituitary Gland Anatomy
- 2. EPIDEMIOLOGY
- 2.1 Incidentalomas
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Pathogenesis
- 3.2 Genetic Syndromes
- 4. CLINICAL FEATURES
- 4.1 Local Mass Effects
- 4.2 Hormonal Hypersecretion Syndromes
- 4.3 Hypothalamic Lesions
- 5. DIFFERENTIAL DIAGNOSIS
- 5.1 Craniopharyngiomas
- 5.2 Other Sellar Masses
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Laboratory Investigation
- 6.2 Imaging
- 7. MANAGEMENT & TREATMENT
- 7.1 Transsphenoidal Surgery
- 7.2 Radiation Therapy
- 7.3 Medical Therapy
- 7.4 Gonadotropin Deficiency Management
- 8. PROGNOSIS & COMPLICATIONS
- 8.1 Surgical Complications
- 9. SPECIAL CONSIDERATIONS
- 9.1 Drug-Induced Hyperprolactinemia
- 10. KEY PEARLS & CLINICAL TRAPS
- 10.1 Diagnostic Clues
- 10.2 Exclusion Criteria
- Flowcharts & Algorithms
- Figures & Illustrations
📋 Figures in This Chapter¶
| # | Type | Description |
|---|---|---|
| 1 | 🔀 Flowchart | Imaging differential diagnosis of sellar masses |
| 2 | 🔀 Flowchart | Management of prolactinoma |
| 3 | 🔀 Flowchart | Management of acromegaly |
| 4 | 🔀 Flowchart | Management of Cushing’s disease |
| 5 | 🔀 Flowchart | Management of a nonfunctioning pituitary mass |
| 1 | 🖼 Figure | Features of acromegaly/gigantism |
| 2 | 🖼 Figure | Transsphenoidal resection of pituitary mass via the endonasal approach |
| 3 | 🖼 Figure | Expanding pituitary mass |
| 4 | 🖼 Figure | Expanding pituitary mass |
| 5 | 🖼 Figure | Expanding pituitary mass |
| 6 | 🖼 Figure | Large invasive prolactinoma successfully treated with cabergoline |
| 7 | 🖼 Figure | Large invasive prolactinoma successfully treated with cabergoline |
| 8 | 🖼 Figure | Large invasive prolactinoma successfully treated with cabergoline |
| 9 | 🖼 Figure | Large invasive prolactinoma successfully treated with cabergoline |
1. DEFINITION & OVERVIEW¶
Pituitary adenomas are benign neoplasms arising from one of the five anterior pituitary cell types. They account for ~15% of all intracranial neoplasms and have a population prevalence of ~80/100,000. At autopsy, up to one-quarter of all pituitary glands harbor an unsuspected microadenoma (<10 mm diameter). Sellar masses may arise from brain, hypothalamic, or pituitary tissues. Each exhibits features related to the lesion location but also unique to the specific etiology.
1.1 Sellar Mass Classification¶
Sellar masses are encountered commonly as incidental findings on MRI, and most are pituitary adenomas (incidentalomas). In the absence of hormone hypersecretion, these small intrasellar lesions can be monitored safely with MRI, which is performed annually and then less often if there is no evidence of further growth. Resection should be considered for incidentally discovered larger macroadenomas, because about one-third become invasive or cause local pressure effects.
1.2 Pituitary Gland Anatomy¶
MRI with gadolinium enhancement allows precise visualization of the pituitary gland with clear delineation of the hypothalamus, pituitary stalk, pituitary tissue and surrounding suprasellar cisterns, cavernous sinuses, sphenoid sinus, and optic chiasm. Pituitary gland height ranges from 6 mm in children to 8 mm in adults; during pregnancy and puberty, the height may reach 10–12 mm. The upper aspect of the adult pituitary is flat or slightly concave, but in adolescent and pregnant individuals, this surface may be convex, reflecting physiologic pituitary enlargement. The stalk should be midline and vertical.
2. EPIDEMIOLOGY¶
Pituitary adenomas are the most common cause of pituitary hormone hypersecretion and hyposecretion syndromes in adults. They account for ~15% of all intracranial neoplasms and have been identified with a population prevalence of ~80/100,000. At autopsy, up to one-quarter of all pituitary glands harbor an unsuspected microadenoma (<10 mm diameter). Similarly, pituitary imaging detects small clinically inapparent pituitary lesions in at least 10% of individuals.
2.1 Incidentalomas¶
Sellar masses are encountered commonly as incidental findings on MRI, and most are pituitary adenomas (incidentalomas). In the absence of hormone hypersecretion, these small intrasellar lesions can be monitored safely with MRI, which is performed annually and then less often if there is no evidence of further growth. Resection should be considered for incidentally discovered larger macroadenomas, because about one-third become invasive or cause local pressure effects.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
Pituitary adenomas are benign neoplasms that arise from one of the five anterior pituitary cell types. The clinical and biochemical phenotypes of pituitary adenomas depend on the cell type from which they are derived. Thus, tumors arising from lactotrope (PRL), somatotrope (GH), corticotrope (ACTH), thyrotrope (TSH), or gonadotrope (LH, FSH) cells hypersecrete their respective hormones. Plurihormonal tumors express various combinations of GH, PRL, TSH, ACTH, or the glycoprotein hormone α or β subunits. Morphologically, these tumors may arise from a single polysecreting cell type or include cells with mixed function within the same tumor.
3.1 Pathogenesis¶
Several etiologic genetic events have been implicated in the development of pituitary tumors. The pathogenesis of sporadic forms of acromegaly has been particularly informative as a model of tumorigenesis. GHRH, after binding to its G protein–coupled somatotrope receptor, uses cyclic adenosine monophosphate (AMP) as a second messenger to stimulate GH secretion and somatotrope proliferation. A subset (~35%) of GH-secreting pituitary tumors contains sporadic mutations in Gα. These mutations attenuate intrinsic GTPase activity, resulting in constitutive elevation of cyclic AMP, Pit-1 induction, and activation of cyclic AMP response element binding protein (CREB), thereby promoting somatotrope cell proliferation and GH secretion. Growth factors may also promote pituitary tumor proliferation. Basic fibroblast growth factor (bFGF) is abundant in the pituitary and stimulates pituitary cell mitogenesis, whereas epidermal growth factor (EGFR) signaling induces both hormone synthesis and cell proliferation. Mutations of USP8 may result in overexpressed EGFR in a subset of ACTH-secreting tumors. Other factors involved in initiation and promotion of pituitary tumors include loss of negative-feedback inhibition (as seen with primary hypothyroidism or hypogonadism) and estrogen-mediated or paracrine angiogenesis. Growth characteristics and neoplastic behavior also may be influenced by activated oncogenes, including RAS and pituitary tumor transforming gene (PTTG), or inactivation of growth suppressor genes, including MEG3.
3.2 Genetic Syndromes¶
Several familial syndromes are associated with pituitary tumors, and the genetic mechanisms for some of them have been unraveled. Multiple endocrine neoplasia (MEN) 1 is an autosomal dominant syndrome characterized primarily by a genetic predisposition to parathyroid, pancreatic islet, and pituitary adenomas. MEN 1 is caused by inactivating germline mutations in MENIN, a constitutively expressed tumor-suppressor gene located on chromosome 11q13. Loss of heterozygosity or a somatic mutation of the remaining normal MENIN allele leads to tumorigenesis. About half of affected patients develop prolactinomas; acromegaly and Cushing's disease are less commonly encountered. Carney complex is characterized by spotty skin pigmentation, myxomas, and endocrine tumors, including testicular, adrenal, and pituitary adenomas. Acromegaly occurs in ~20% of these patients. A subset of patients has mutations in the R1α regulatory subunit of protein kinase A (PRKAR1A). Familial acromegaly is a rare disorder in which family members may manifest either acromegaly or gigantism. A subset of families with a predisposition for familial pituitary tumors, especially acromegaly, has been found to harbor germline mutations in the AIP gene, which encodes the aryl hydrocarbon receptor interacting protein.
Table 1 — TABLE 392-4 Familial Pituitary Tumor Syndromes¶
| GENE MUTATED | CLINICAL FEATURES |
|---|---|
| Multiple endocrine neoplasia 1 (MEN 1) (11q13) | Hyperparathyroidism; Pancreatic neuroendocrine tumors; Foregut carcinoids; Adrenal adenomas; Skin lesions; Pituitary adenomas (40%) |
| Multiple endocrine neoplasia 4 (MEN 4) (12p13) | Pituitary adenomas; Other tumors |
| Carney complex (17q23-24) | Pituitary hyperplasia and adenomas (10%); Atrial myxomas; Schwannomas; Adrenal hyperplasia; Lentigines |
| Familial pituitary adenomas (11q13.2) | Acromegaly/gigantism (~15% of afflicted families) |
4. CLINICAL FEATURES¶
Clinical manifestations of sellar lesions vary, depending on the anatomic location of the mass and the direction of its extension. Headaches are common features of small intrasellar tumors, even with no demonstrable suprasellar extension. Because of the confined nature of the pituitary, small changes in intrasellar pressure stretch the dural plate; however, headache severity correlates poorly with adenoma size or extension. Suprasellar extension can lead to visual loss by several mechanisms, the most common being compression of the optic chiasm. Rarely, direct invasion of the optic nerves or obstruction of cerebrospinal fluid (CSF) flow leading to secondary visual disturbances can occur. Pitu-itary stalk compression by a hormonally active or inactive intrasellar mass may compress the portal vessels, disrupting pituitary access to hypothalamic hormones and dopamine; this results in early hyperprolactinemia and later concurrent loss of other pituitary hormones. This "stalk section" phenomenon may also be caused by trauma, whiplash injury with posterior clinoid stalk compression, or skull base fractures.
4.1 Local Mass Effects¶
Local mass effects include headaches, visual field defects, cranial nerve palsies, and hydrocephalus. Lateral mass invasion may impinge on the cavernous sinus and compress its neural contents, leading to cranial nerve III, IV, and VI palsies as well as effects on the ophthalmic and maxillary branches of the fifth cranial nerve. Patients may present with diplopia, ptosis, ophthalmoplegia, and decreased facial sensation, depending on the extent of neural damage. Extension into the sphenoid sinus indicates that the pituitary mass has eroded through the sellar floor. Aggressive tumors rarely invade the palate roof and cause nasopharyngeal obstruction, infection, and CSF leakage. Temporal and frontal lobe involvement may rarely lead to uncinate seizures, personality disorders, and anosmia. Direct hypothalamic encroachment by an invasive pituitary mass may cause important metabolic sequelae, including precocious puberty or hypogonadism, arginine vasopressin deficiency (AVP-D), sleep disturbances, dysthermia, and appetite disorders.
Table 2 — TABLE 392-1 Features of Sellar Mass Lesions¶
| IMPACTED STRUCTURE | CLINICAL IMPACT |
|---|---|
| Pituitary | Hypogonadism; Hypothyroidism; Growth failure, adult growth hormone deficiency; Hypoadrenalism; Hyperprolactinemia (stalk compression) |
| Optic chiasm | Bitemporal hemianopia; Superior or bitemporal field defect; Scotoma; Blindness; Loss of red perception |
| Hypothalamus | Temperature dysregulation; Appetite and thirst disorders; Obesity; Sleep disorders; Behavioral dysfunction; Autonomic dysfunction |
| Cavernous sinus | Ophthalmoplegia with or without ptosis or diplopia; Facial numbness |
| Frontal lobe | Personality disorder; Anosmia |
| Brain | Headache; Hydrocephalus; Psychosis; Dementia; Laughing seizures |
4.2 Hormonal Hypersecretion Syndromes¶
Pituitary adenomas are the most common cause of pituitary hormone hypersecretion and hyposecretion syndromes in adults. They account for ~15% of all intracranial neoplasms and have been identified with a population prevalence of ~80/100,000. Hormonally active tumors are characterized by autonomous hormone secretion with diminished feedback responsiveness to physiologic inhibitory pathways. Hormone production does not always correlate with tumor size. Small hormone-secreting adenomas may cause significant clinical perturbations, whereas larger adenomas that produce less hormone may be clinically silent and remain undiagnosed (if no central compressive effects occur). About one-third of all adenomas are clinically nonfunctioning and produce no distinct clinical hypersecretory syndrome. Most of them arise from gonadotrope cells and may secrete small amounts of α- and β-glycoprotein hormone subunits or, very rarely, intact circulating gonadotropins. True pituitary carcinomas with documented extracranial metastases are exceedingly rare.
Table 3 — TABLE 392-3 Classification of Pituitary Adenomas¶
| ADENOMA CELL ORIGIN | HORMONE PRODUCT | CLINICAL SYNDROME |
|---|---|---|
| Lactotrope | PRL | Hypogonadism, galactorrhea |
| Gonadotrope | FSH, LH, subunits | Silent/nonfunctioning, ovarian hyperstimulation, hypogonadism |
| Somatotrope | GH | Acromegaly/gigantism |
| Corticotrope | ACTH/none | Cushing's disease or silent |
| Mixed lactotrope and somatotrope | GH, PRL | Acromegaly, hypogonadism, galactorrhea |
| Any | Mixed | Mixed |
| Acidophil stem cell | PRL, GH | Hypogonadism, galactorrhea, acromegaly |
| Mammosomatotrope | PRL, GH | Hypogonadism, galactorrhea, acromegaly |
| Thyrotrope | TSH | Thyrotoxicosis |
| None | None | Hypopituitarism/none |
| Oncocytoma | None | Hypopituitarism/none |
4.3 Hypothalamic Lesions¶
Lesions involving the anterior and preoptic hypothalamic regions cause paradoxical vasoconstriction, tachycardia, and hyperthermia. Acute hyperthermia usually is due to a hemorrhagic insult, but poikilothermia may also occur. Central disorders of thermoregulation result from posterior hypothalamic damage. The periodic hypothermia syndrome is characterized by episodic attacks of rectal temperatures <30°C (86°F), sweating, vasodilation, vomiting, and bradycardia. Damage to the ventromedial hypothalamic nuclei by craniopharyngiomas, hypothalamic trauma, or inflammatory disorders may be associated with hyperphagia and obesity. This region appears to contain an energy-satiety center where melanocortin receptors are influenced by leptin, insulin, pro-opiomelanocortin (POMC) products, and gastrointestinal peptides. Polydipsia and hypodipsia are associated with damage to central osmoreceptors located in preoptic nuclei. Slow-growing hypothalamic lesions can cause increased somnolence and disturbed sleep cycles as well as obesity, hypothermia, and emotional outbursts. Lesions of the central hypothalamus may stimulate sympathetic neurons, leading to elevated serum catecholamine and cortisol levels. These patients are predisposed to cardiac arrhythmias, hypertension, and gastric erosions.
5. DIFFERENTIAL DIAGNOSIS¶
Sellar masses may arise from brain, hypothalamic, or pituitary tissues. Each exhibit features related to the lesion location but also unique to the specific etiology. Unique MRI characteristics inform the differential diagnosis of pituitary masses. Lesions involving the anterior and preoptic hypothalamic regions cause paradoxical vasoconstriction, tachycardia, and hyperthermia. Acute hyperthermia usually is due to a hemorrhagic insult, but poikilothermia may also occur. Central disorders of thermoregulation result from posterior hypothalamic damage. The periodic hypothermia syndrome is characterized by episodic attacks of rectal temperatures <30°C (86°F), sweating, vasodilation, vomiting, and bradycardia. Damage to the ventromedial hypothalamic nuclei by craniopharyngiomas, hypothalamic trauma, or inflammatory disorders may be associated with hyperphagia and obesity. This region appears to contain an energy-satiety center where melanocortin receptors are influenced by leptin, insulin, pro-opiomelanocortin (POMC) products, and gastrointestinal peptides. Polydipsia and hypodipsia are associated with damage to central osmoreceptors located in preoptic nuclei. Slow-growing hypothalamic lesions can cause increased somnolence and disturbed sleep cycles as well as obesity, hypothermia, and emotional outbursts. Lesions of the central hypothalamus may stimulate sympathetic neurons, leading to elevated serum catecholamine and cortisol levels. These patients are predisposed to cardiac arrhythmias, hypertension, and gastric erosions.
5.1 Craniopharyngiomas¶
Craniopharyngiomas are benign, suprasellar cystic masses that present with headaches, visual field deficits, and variable degrees of hypopituitarism. They are derived from Rathke's pouch and arise near the pituitary stalk, commonly extending into the suprasellar cistern. Craniopharyngiomas are often large, cystic, and locally invasive. Many are partially calcified, exhibiting a characteristic appearance on skull x-ray and CT images. More than half of all patients present before age 20, usually with signs of increased intracranial pressure, including headache, vomiting, papilledema, and hydrocephalus. Associated symptoms include visual field abnormalities, personality changes and cognitive deterioration, cranial nerve damage, sleep difficulties, and weight gain accompanied by features of the metabolic syndrome. Hypopituitarism is documented in ~90%, and AVP-D occurs in ~10% of patients. About half of affected children present with growth retardation. MRI is generally superior to CT for evaluating cystic structure and tissue components of craniopharyngiomas. CT is useful to define calcifications and evaluate invasion into surrounding bony structures and sinuses.
5.2 Other Sellar Masses¶
Rathke's cysts are small (<5 mm) cysts entrapped by squamous epithelium and are found in ~20% of individuals at autopsy. Although Rathke's cysts do not usually grow and are often diagnosed incidentally, about a third present in adulthood with compressive symptoms, AVP-D, and hyperprolactinemia due to stalk compression. Rarely, hydrocephalus develops. The diagnosis is suggested preoperatively by visualizing the cyst wall on MRI, which distinguishes these lesions from craniopharyngiomas. Cyst contents range from CSF-like fluid to mucoid material. Arachnoid cysts are rare and generate an MRI image that is isointense with CSF. Sellar chordomas usually present with bony clival erosion, local invasiveness, and, on occasion, calcification. Normal pituitary tissue may be visible on MRI, distinguishing chordomas from aggressive pituitary adenomas. Mucinous material may be obtained by fine-needle aspiration. They may be diagnosed by careful immunocytochemistry of specific histology. Meningiomas arising in the sellar region may be difficult to distinguish from nonfunctioning pituitary adenomas. Meningiomas typically enhance on MRI and may show evidence of calcification or bony erosion. Meningiomas may cause compressive symptoms. Histiocytosis X includes a variety of syndromes associated with foci of eosinophilic granulomas. AVP-D, exophthalmos, and punched-out lytic bone lesions (Hand-Schüller-Christian disease) are associated with granulomatous lesions visible on MRI, as well as a characteristic axillary skin rash. Rarely, the pituitary stalk may be involved. Pituitary metastases occur in ~3% of cancer patients. Bloodborne metastatic deposits are found almost exclusively in the posterior pituitary. Accordingly, AVP-D can be a presenting feature of lung, gastrointestinal, breast, and other pituitary metastases. About half of pituitary metastases originate from breast cancer; ~25% of patients with metastatic breast cancer have such deposits. Rarely, pituitary stalk involvement results in anterior pituitary insufficiency. The MRI diagnosis of a metastatic lesion may be difficult to distinguish from an aggressive pituitary adenoma; the diagnosis may require histologic examination of excised tumor tissue. Primary or metastatic lymphoma, leukemias, and plasmacytomas also occur within the sella. Hypothalamic hamartomas and gangliocytomas may arise from astrocytes, oligodendrocytes, and neurons with varying degrees of differentiation. These tumors may overexpress hypothalamic neuropeptides, including gonadotropin-releasing hormone (GnRH), growth hormone–releasing hormone (GHRH), and corticotropin-releasing hormone (CRH). With GnRH-producing tumors, children present with precocious puberty, psychomotor delay, and laughing-associated seizures. Medical treatment of GnRH-producing hamartomas with long-acting GnRH analogues effectively suppresses gonadotropin secretion and controls premature pubertal development. Rarely, hamartomas also are associated with craniofacial abnormalities; imperforate anus; cardiac, renal, and lung disorders; and pituitary failure as features of Pallister-Hall syndrome, which is caused by mutations in the carboxy terminus of the GLI3 gene. Hypothalamic hamartomas are often contiguous with the pituitary, and preoperative MRI diagnosis may not be possible. Histologic evidence of hypothalamic neurons in tissue resected at transsphenoidal surgery may be the first indication of a primary hypothalamic lesion. Hypothalamic gliomas and optic gliomas occur mainly in childhood and usually present with visual loss. Adults have more aggressive tumors; about a third are associated with neurofibromatosis. Brain germ cell tumors may arise within the sellar region. They include dysgerminomas, which frequently are associated with AVP-D and visual loss. They rarely metastasize. Germinomas, embryonal carcinomas, teratomas, and choriocarcinomas may arise in the parasellar region and produce human chorionic gonadotropin (hCG). These germ cell tumors present with precocious puberty, AVP-D, visual field defects, and thirst disorders. Many patients are GH deficient with short stature.
6. INVESTIGATIONS & DIAGNOSIS¶
MRI examination of the sellar region and assessment of other pituitary functions usually are indicated in patients with documented central hypogonadism. When a pituitary adenoma is suspected based on MRI, initial hormonal evaluation usually includes (1) basal prolactin (PRL); (2) insulin-like growth factor (IGF)-1; (3) 24-h urinary free cortisol (UFC) and/or overnight oral dexamethasone (1 mg) suppression test; (4) α subunit, follicle-stimulating hormone (FSH), and luteinizing hormone (LH); and (5) thyroid function tests. Additional hormonal evaluation may be indicated based on the results of these tests. Pending more detailed assessment of hypopituitarism, a menstrual history, measurement of testosterone and 8 A.M. cortisol levels, and thyroid function tests usually identify patients with pituitary hormone deficiencies that require hormone replacement before further testing or surgery. Immunohistochemical staining of pituitary tumor specimens obtained at transsphenoidal surgery for hormones as well as cell-type specific transcription factors confirms clinical and laboratory studies and provides a histologic diagnosis and confirms clinical and laboratory studies and provides a histologic diagnosis and confirms clinical and laboratory studies and provides a histologic diagnosis when hormone studies are equivocal and in cases of clinically nonfunctioning tumors.
6.1 Laboratory Investigation¶
Central hypogonadism is associated with low or inappropriately normal serum gonadotropin levels in the setting of low sex hormone concentrations (testosterone in men, estradiol in women). Because gonadotropin secretion is pulsatile, valid assessments may require repeated measurements or the use of pooled serum samples. Men have reduced sperm counts. Intravenous GnRH (100 μg) stimulates gonadotropes to secrete LH (which peaks within 30 min) and FSH (which plateaus during the ensuing 60 min). Normal responses vary according to menstrual cycle stage, age, and sex of the patient. Generally, LH levels increase about threefold, whereas FSH responses are less pronounced. In the setting of gonadotropin deficiency, a normal gonadotropin response to GnRH indicates intact pituitary gonadotrope function and suggests a hypothalamic abnormality. An absent response, however, does not reliably distinguish pituitary from hypothalamic causes of hypogonadism. For this reason, GnRH testing usually adds little to the information gained from baseline evaluation of the hypothalamic-pituitary-gonadotrope axis except in cases of isolated GnRH deficiency (e.g., Kallmann syndrome).
Table 4 — TABLE 392-2 Screening Tests for Functional Pituitary Adenomas¶
| TEST | COMMENTS |
|---|---|
| Acromegaly | Serum IGF-1; Interpret IGF-1 relative to age- and sex-matched controls; Normal subjects should suppress growth hormone to <1 μg/L; Oral glucose tolerance test with GH obtained at 0, 30, and 60 min |
| Prolactinoma | Serum PRL; Exclude medications; MRI of the sella should be ordered if PRL is elevated |
| Cushing's disease | 24-h urinary free cortisol; Ensure urine collection is total and accurate; Dexamethasone (1 mg) at 11 P.m. and fasting plasma cortisol measured at 8 a.m.; Normal subjects suppress to <5 μg/dL; Late night salivary cortisol; ACTH assay; CRH stimulation test with measurements of cortisol and ACTH from peripheral and/or petrosal sinus blood; The CRH test is used primarily to distinguish pituitary adenomas from ectopic ACTH sources |
| Gonadotropinoma | Baseline FSH, LH, free α subunit, ovarian hyperstimulation, estrogen (females), testosterone (males); TRH stimulation test with assays for LH, FSH, free α subunit, free LHβ, free FSHβ subunits |
| TSH-producing adenoma | Free T, free T4, TSH, free α subunit; Key feature is an inappropriately normal or high TSH in the setting of elevated free T and T4 |
6.2 Imaging¶
MRI with gadolinium enhancement for pituitary visualization, new advances in transsphenoidal surgery and in stereotactic radiotherapy, and novel therapeutic agents have improved pituitary tumor management. MRI with gadolinium enhancement allows precise visualization of the pituitary gland with clear delineation of the hypothalamus, pituitary stalk, pituitary tissue and surrounding suprasellar cisterns, cavernous sinuses, sphenoid sinus, and optic chiasm. Pituitary gland height ranges from 6 mm in children to 8 mm in adults; during pregnancy and puberty, the height may reach 10–12 mm. The upper aspect of the adult pituitary is flat or slightly concave, but in adolescent and pregnant individuals, this surface may be convex, reflecting physiologic pituitary enlargement. The stalk should be midline and vertical. Anterior pituitary gland soft tissue consistency is slightly heterogeneous on MRI, and signal intensity resembles that of brain matter on T1-weighted imaging. Adenoma density is usually lower than that of surrounding normal tissue on T1-weighted imaging, and the signal intensity increases with T2-weighted images. Computed tomography (CT) scan is reserved to define the extent of bony erosion or the presence of calcification.
7. MANAGEMENT & TREATMENT¶
Successful management of sellar masses requires accurate diagnosis as well as selection of optimal therapeutic modalities. Most pituitary tumors are benign and slow growing. Clinical features result from local mass effects and hormonal hyper- or hyposecretion syndromes caused directly by the adenoma or occurring as a consequence of treatment. Thus, lifelong management and follow-up are necessary for these patients. The goals of pituitary tumor treatment include normalization of excess pituitary secretion, amelioration of symptoms and signs of hormonal hypersecretion syndromes, and shrinkage or ablation of large tumor masses with relief of adjacent structure compression. Residual anterior pituitary function should be preserved during treatment and sometimes can be restored by removing the tumor mass. Ideally, adenoma recurrence should be prevented.
7.1 Transsphenoidal Surgery¶
Transsphenoidal resection is the desired surgical approach for pituitary tumors, except for the rare invasive suprasellar mass surrounding the frontal or middle fossa or the optic nerves or invading posteriorly behind the clivus, which may require transcranial approaches. Intraoperative microscopy facilitates visual distinction between adenomatous and normal pituitary tissue as well as microdissection of small tumors that may not be visible by MRI. Endoscopic techniques with three-dimensional intraoperative localization enable better visualization and access to tumor tissue. Transsphenoidal surgery also avoids cranial invasion and manipulation of brain tissue required by subfrontal surgical approaches. Individual surgical experience is a major determinant of outcome efficacy with these techniques. In addition to correction of hormonal hypersecretion, pituitary surgery is indicated for mass lesions that impinge on surrounding structures. Surgical decompression and resection are required for an expanding pituitary mass, which may be asymptomatic or accompanied by persistent headache, progressive visual field defects, cranial nerve palsies, hydrocephalus, and, occasionally, intrapituitary hemorrhage and apoplexy. Transsphenoidal surgery rarely is used for pituitary tissue biopsy to establish a histologic diagnosis. Whenever possible, the pituitary mass lesion should be selectively excised; normal pituitary tissue should be manipulated or resected only when critical for effective mass dissection. Non-selective hemihypophysectomy or total hypophysectomy may be indicated if no hypersecreting mass lesion is clearly discernible, multifocal lesions are present, or the remaining nontumorous pituitary tissue is obviously necrotic. This strategy, however, increases the likelihood of postoperative hypopituitarism and the need for lifelong hormone replacement. Preoperative mass effects, including visual field defects and compromised pituitary function, may be reversed by surgery, particularly when the deficits are not long-standing. For large and invasive tumors, it is necessary to determine the optimal balance between maximal tumor resection and preservation of anterior pituitary hormonal function, especially for preserving growth and reproductive function in younger patients. Tumor invasion outside the sella is rarely amenable to surgical cure, and the surgeon must judge the risk-versus-benefit ratio of extensive tumor resection.
7.2 Radiation Therapy¶
Radiation is used either as a primary therapy for pituitary or parasellar masses or, more commonly, as an adjunct to surgery or medical therapy. Focused megavoltage irradiation is achieved by precise MRI localization, using a high-voltage linear accelerator and accurate isocentric rotational arcing. A major determinant of accurate irradiation is reproduction of the patient's head position during multiple visits and maintenance of absolute head immobility. A total of <50 Gy (5000 rad) is given as 180 cGy (180 rad) fractions divided over ~6 weeks. Stereotactic radiosurgery delivers a large single high-energy dose from a cobalt-60 source (Gamma Knife), linear accelerator, or cyclotron. Long-term effects of Gamma Knife surgery appear to be similar to those encountered with conventional radiation. Proton beam therapy is available in some centers and provides concentrated radiation doses within a localized region. Side effects in the short term, radiation may cause transient nausea and weakness. Alopecia and loss of taste and smell may be more long-lasting. Failure of pituitary hormone synthesis is common in patients who have undergone head and neck or pituitary-directed irradiation. More than 50% of patients develop loss of GH, ACTH, TSH, and/or gonadotropin secretion within 10 years, usually due to hypothalamic damage. Lifelong follow-up with testing of anterior pituitary hormone reserve is therefore required after radiation treatment. Optic nerve damage with impaired vision due to optic neuritis is reported in ~2% of patients who undergo pituitary irradiation. Cranial nerve damage is uncommon now that radiation doses are <2 Gy (200 rad) at any one treatment session and the maximum dose is <50 Gy (5000 rad). The use of stereotactic radiotherapy reduces the risk of damage to adjacent structures. Conventional radiotherapy for pituitary tumors has been associated with adverse mortality rates, mainly from cerebrovascular disease. The cumulative risk of developing a secondary tumor after conventional radiation is 1.3% after 10 years and 1.9% after 20 years.
7.3 Medical Therapy¶
Medical therapy for pituitary tumors is highly specific and depends on tumor type. For prolactinomas, dopamine agonists are the treatment of choice. For acromegaly, somatostatin receptor ligands (SRLs) and a GH receptor antagonist are indicated. For TSH-secreting tumors, SRLs and occasionally dopamine agonists are indicated. ACTH-secreting tumors may respond to SRLs, and adrenal-directed therapy may also be of benefit. Nonfunctioning tumors are generally not responsive to medications and require surgery and/or irradiation. Hypothalamic, Pituitary, and Other Sellar Masses: Sellar masses may arise from brain, hypothalamic, or pituitary tissues. Each exhibit features related to the lesion location but also unique to the specific etiology. Unique MRI characteristics inform the differential diagnosis of pituitary masses. Lesions involving the anterior and preoptic hypothalamic regions cause paradoxical vasoconstriction, tachycardia, and hyperthermia. Acute hyperthermia usually is due to a hemorrhagic insult, but poikilothermia may also occur. Central disorders of thermoregulation result from posterior hypothalamic damage. The periodic hypothermia syndrome is characterized by episodic attacks of rectal temperatures <30°C (86°F), sweating, vasodilation, vomiting, and bradycardia. Damage to the ventromedial hypothalamic nuclei by craniopharyngiomas, hypothalamic trauma, or inflammatory disorders may be associated with hyperphagia and obesity. This region appears to contain an energy-satiety center where melanocortin receptors are influenced by leptin, insulin, pro-opiomelanocortin (POMC) products, and gastrointestinal peptides. Polydipsia and hypodipsia are associated with damage to central osmoreceptors located in preoptic nuclei. Slow-growing hypothalamic lesions can cause increased somnolence and disturbed sleep cycles as well as obesity, hypothermia, and emotional outbursts. Lesions of the central hypothalamus may stimulate sympathetic neurons, leading to elevated serum catecholamine and cortisol levels. These patients are predisposed to cardiac arrhythmias, hypertension, and gastric erosions.
7.4 Gonadotropin Deficiency Management¶
In males, testosterone replacement is necessary to achieve and maintain normal growth and development of the external genitalia, secondary sex characteristics, male sexual behavior, and androgenic effects, including maintenance of muscle function and bone mass. Testosterone may be administered by intramuscular injections every 1–4 weeks or by using skin patches or testosterone gels. Gonadotropin injections (hCG or human menopausal gonadotropin [hMG]) over 12–18 months are used to restore fertility. Pulsatile GnRH therapy (25–150 ng/kg every 2 h), administered by a subcutaneous infusion pump, is also effective for treatment of hypothalamic hypogonadism when fertility is desired. In premenopausal women, cyclical replacement of estrogen and progesterone maintains secondary sexual characteristics and integrity of genitourinary tract mucosa and prevents premature osteoporosis. Gonadotropin therapy is used for ovulation induction. Follicular growth and maturation are initiated using hMG or recombinant FSH; hCG or human luteinizing hormone is subsequently injected to induce ovulation. As in men, pulsatile GnRH therapy can be used to treat hypothalamic causes of gonadotropin deficiency.
8. PROGNOSIS & COMPLICATIONS¶
Most pituitary tumors are benign and slow growing. Clinical features result from local mass effects and hormonal hyper- or hyposecretion syndromes caused directly by the adenoma or occurring as a consequence of treatment. Thus, lifelong management and follow-up are necessary for these patients. The goals of pituitary tumor treatment include normalization of excess pituitary secretion, amelioration of symptoms and signs of hormonal hypersecretion syndromes, and shrinkage or ablation of large tumor masses with relief of adjacent structure compression. Residual anterior pituitary function should be preserved during treatment and sometimes can be restored by removing the tumor mass. Ideally, adenoma recurrence should be prevented. For large and invasive tumors, it is necessary to determine the optimal balance between maximal tumor resection and preservation of anterior pituitary hormonal function, especially for preserving growth and reproductive function in younger patients. Tumor invasion outside the sella is rarely amenable to surgical cure, and the surgeon must judge the risk-versus-benefit ratio of extensive tumor resection. For craniopharyngiomas, surgery alone is curative in less than half of patients because of recurrences due to adherence to vital structures or because of small tumor deposits in the hypothalamus or brain parenchyma. In the absence of radiotherapy, ~75% of craniopharyngiomas recur, and 10-year survival is <50%. In patients with incomplete resection, radiotherapy improves 10-year survival to 70–90% but is associated with increased risk of secondary malignancies. Most patients require lifelong pituitary hormone replacement. As some craniopharyngiomas (particularly papillary) are associated with activated BRAF V600E mutations, use of BRAF inhibitors (dabrafenib or vemurafenib) either alone or in combination with MEK inhibitors (trametinib or cobimetinib) has resulted in long-term growth responses in some patients.
8.1 Surgical Complications¶
Side effects include transient AVP-D and hypopituitarism occur in up to 20% of patients. Permanent AVP-D, cranial nerve damage, nasal septal perforation, or visual disturbances may be encountered in up to 10% of patients. CSF leaks occur in 4% of patients. Less common complications include carotid artery injury, loss of vision, hypothalamic damage, and meningitis. Permanent side effects are rare after surgery for microadenomas. Operative mortality rate is ~1%. Transient AVP-D and hypopituitarism occur in up to 20% of patients. Permanent AVP-D, cranial nerve damage, nasal septal perforation, or visual disturbances may be encountered in up to 10% of patients. CSF leaks occur in 4% of patients. Less common complications include carotid artery injury, loss of vision, hypothalamic damage, and meningitis.
9. SPECIAL CONSIDERATIONS¶
Pregnancy and lactation are the important physiologic causes of hyperprolactinemia. Sleep-associated hyperprolactinemia reverts to normal within an hour of awakening. Nipple stimulation and sexual orgasm also may increase PRL. Chest wall stimulation or trauma (including chest surgery and herpes zoster) invokes the reflex suckling arc with resultant hyperprolactinemia. Chronic renal failure elevates PRL by decreasing peripheral clearance. Primary hypothyroidism is associated with mild hyperprolactinemia, probably because of compensatory TRH secretion. Mutation of the PRL receptor is a rare cause of hyperprolactinemia. Lesions of the hypothalamic-pituitary region that disrupt hypothalamic dopamine synthesis, portal vessel delivery, or lactotrope responses are associated with hyperprolactinemia. Thus, hypothalamic tumors, cysts, infiltrative disorders, and radiation-induced damage cause elevated PRL levels, usually in the range of 30–100 μg/L. Plurihormonal adenomas (including GH and ACTH tumors) may hypersecrete PRL directly. Pituitary masses, including clinically nonfunctioning pituitary tumors, may compress the pituitary stalk to cause hyperprolactinemia. Drug-induced inhibition or disruption of dopaminergic receptor function is a common cause of hyperprolactinemia. Thus, antipsychotics and antidepressants are a relatively common cause of mild hyperprolactinemia. Most patients receiving risperidone have elevated PRL levels, sometimes exceeding 200 μg/L. Methyldopa inhibits dopamine synthesis, and verapamil blocks dopamine release, also leading to hyperprolactinemia. Hormonal agents that induce PRL include estrogens and thyrotropin-releasing hormone (TRH).
9.1 Drug-Induced Hyperprolactinemia¶
Drug-induced inhibition or disruption of dopaminergic receptor function is a common cause of hyperprolactinemia. Thus, antipsychotics and antidepressants are a relatively common cause of mild hyperprolactinemia. Most patients receiving risperidone have elevated PRL levels, sometimes exceeding 200 μg/L. Methyldopa inhibits dopamine synthesis, and verapamil blocks dopamine release, also leading to hyperprolactinemia. Hormonal agents that induce PRL include estrogens and thyrotropin-releasing hormone (TRH).
10. KEY PEARLS & CLINICAL TRAPS¶
Hyperprolactinemia is the most common pituitary hormone hypersecretion syndrome in both men and women. PRL-secreting pituitary adenomas (prolactinomas) are the most common cause of PRL levels >200 μg/L (see below). Less pronounced PRL elevation can also be seen with microprolactinomas but is more commonly caused by drugs, pituitary stalk compression, hypothyroidism, or renal failure. Pregnancy and lactation are the important physiologic causes of hyperprolactinemia. Sleep-associated hyperprolactinemia reverts to normal within an hour of awakening. Nipple stimulation and sexual orgasm also may increase PRL. Chest wall stimulation or trauma (including chest surgery and herpes zoster) invokes the reflex suckling arc with resultant hyperprolactinemia. Chronic renal failure elevates PRL by decreasing peripheral clearance. Primary hypothyroidism is associated with mild hyperprolactinemia, probably because of compensatory TRH secretion. Mutation of the PRL receptor is a rare cause of hyperprolactinemia. Lesions of the hypothalamic-pituitary region that disrupt hypothalamic dopamine synthesis, portal vessel delivery, or lactotrope responses are associated with hyperprolactinemia. Thus, hypothalamic tumors, cysts, infiltrative disorders, and radiation-induced damage cause elevated PRL levels, usually in the range of 30–100 μg/L. Plurihormonal adenomas (including GH and ACTH tumors) may hypersecrete PRL directly. Pituitary masses, including clinically nonfunctioning pituitary tumors, may compress the pituitary stalk to cause hyperprolactinemia. Drug-induced inhibition or disruption of dopaminergic receptor function is a common cause of hyperprolactinemia. Thus, antipsychotics and antidepressants are a relatively common cause of mild hyperprolactinemia. Most patients receiving risperidone have elevated PRL levels, sometimes exceeding 200 μg/L. Methyldopa inhibits dopamine synthesis, and verapamil blocks dopamine release, also leading to hyperprolactinemia. Hormonal agents that induce PRL include estrogens and thyrotropin-releasing hormone (TRH).
10.1 Diagnostic Clues¶
Bitemporal hemianopia, often more pronounced superiorly, is observed classically. It occurs because nasal ganglion cell fibers, which cross in the optic chiasm, are especially vulnerable to compression of the ventral optic chiasm. Occasionally, homonymous hemianopia occurs from postchiasmal compression or monocular temporal field loss from prechiasmal compression. Invasion of the cavernous sinus can produce diplopia from ocular motor nerve palsy. Early diagnosis reduces the risk of optic atrophy, vision loss, or eye misalignment. Loss of red perception is a specific finding. The presenting clinical features of functional pituitary adenomas (e.g., acromegaly, prolactinoma, or Cushing's disease) should guide the laboratory studies. However, for a sellar mass with no obvious clinical features of hormone excess, laboratory studies are geared toward determining the nature of the tumor and assessing the possible presence of hypopituitarism.
10.2 Exclusion Criteria¶
The diagnosis of idiopathic hyperprolactinemia is made by exclusion of known causes of hyperprolactinemia in the setting of a normal MRI of the sella. Drug-induced hyperprolactinemia is a common cause. Primary hypothyroidism is associated with mild hyperprolactinemia. Chronic renal failure elevates PRL by decreasing peripheral clearance. Lesions of the hypothalamic-pituitary region that disrupt hypothalamic dopamine synthesis, portal vessel delivery, or lactotrope responses are associated with hyperprolactinemia. Thus, hypothalamic tumors, cysts, infiltrative disorders, and radiation-induced damage cause elevated PRL levels, usually in the range of 30–100 μg/L. Plurihormonal adenomas (including GH and ACTH tumors) may hypersecrete PRL directly. Pituitary masses, including clinically nonfunctioning pituitary tumors, may compress the pituitary stalk to cause hyperprolactinemia.
Flowcharts & Algorithms¶
Reproduced from Harrison's 22nd Edition.
Flowchart 1¶
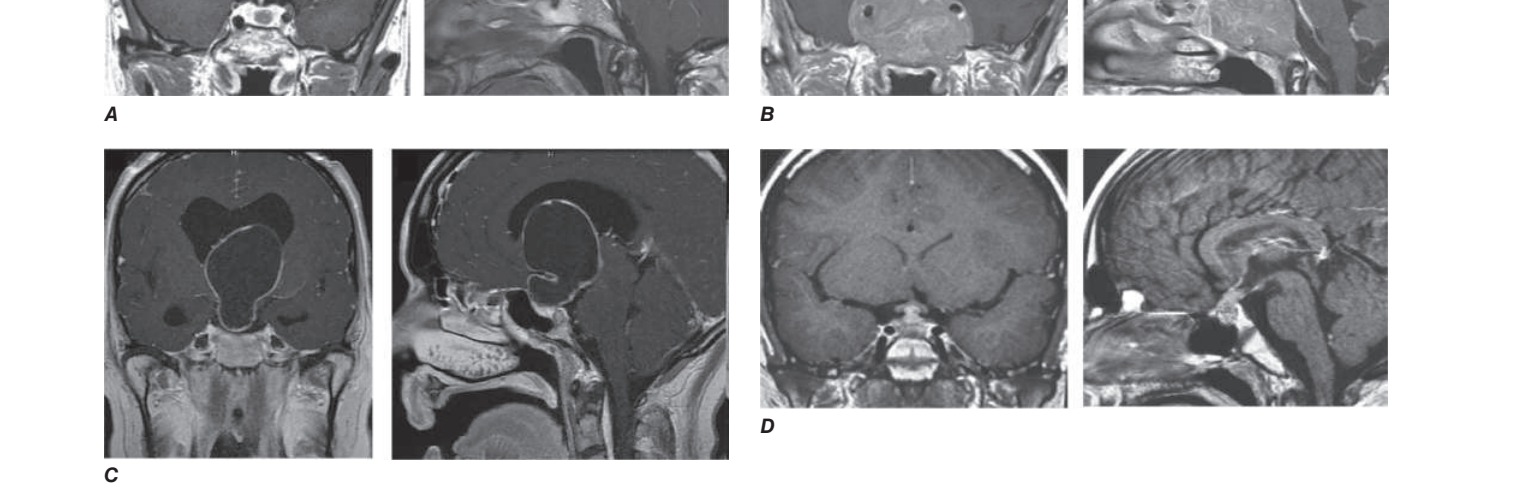
Caption: FIGURE 392-4 Imaging differential diagnosis of sellar masses. A. Microadenoma. B. (A, B, D: Used with permission from Vivien Bonert, MD. C: Reproduced with treatment and follow-up. Horm Res 69:193, 2008.)
Flowchart 2¶

Caption: FIGURE 392-5 Management of prolactinoma. MRI, magnetic resonance imaging; PRL,
Flowchart 3¶

Caption: FIGURE 392-8 Management of acromegaly. aIf curative surgery is not feasible. insulin-like growth factor; SRL, somatostatin receptor ligand (injectable or oral GH RECEPTOR ANTAGONIST Pegvisomant antagonizes endogenous GH action by blocking peripheral GH binding to its receptor. Consequently, serum IGF-1 levels are suppressed, reducing the deleterious effects of excess endogenous GH. Pegvisomant is administered by daily subcu- taneous injection (10–30 mg) and normalizes IGF-1 in ~70% of patients. GH levels, however, remain elevated as the drug does not
Flowchart 4¶

Caption: FIGURE 392-9 Management of Cushing’s disease. ACTH, adrenocorticotropin hormone; MRI, magnetic resonance imaging; ∗, Not usually required.
Flowchart 5¶

Caption: FIGURE 392-10 Management of a nonfunctioning pituitary mass. MRI, magnetic
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 392-7 Features of acromegaly/gigantism. A 22-year-old man with gigantism due height and prognathism (A) and enlarged hand (B) and foot (C) of the affected twin are with permission from RF Gagel, IE McCutcheon. Images in clinical medicine. Pituitary treatment for preoperative shrinkage of large invasive macroadeno- mas, immediate relief of debilitating symptoms, and reduction of GH hypersecretion; in frail patients experiencing morbidity; and in patients who decline surgery or when surgery fails to achieve bio- chemical control. Irradiation or repeat surgery may be required for — FIGURE 392-1 Expanding pituitary mass. Pituitary mass expansion may (A) impinge vital soft tissue structures and (B) invade the sphenoid sinus.
Figure 2¶

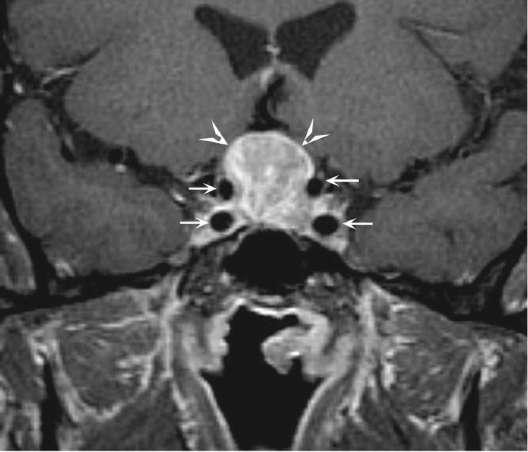
Caption: FIGURE 392-3 Transsphenoidal resection of pituitary mass via the endonasal approach. — FIGURE 392-2 Pituitary adenoma. Coronal T1-weighted postcontrast magnetic resonance image shows a homogeneously enhancing mass (arrowheads) in the sella turcica and suprasellar region compatible with a pituitary adenoma; the small arrows outline the carotid arteries.
Figure 3¶

Caption: FIGURE 392-1 Expanding pituitary mass. Pituitary mass expansion may (A) impinge permission from P Cappabianca et al: Size does not matter. The intrigue of giant — FIGURE 392-3 Transsphenoidal resection of pituitary mass via the endonasal approach.
Figure 4¶

Caption: FIGURE 392-1 Expanding pituitary mass. Pituitary mass expansion may (A) impinge permission from P Cappabianca et al: Size does not matter. The intrigue of giant — FIGURE 392-4 Imaging differential diagnosis of sellar masses. A. Microadenoma. B. Macroadenoma. C. Craniopharyngioma. D. Hypophysitis with stalk thickening.
Figure 5¶
Caption: FIGURE 392-1 Expanding pituitary mass. Pituitary mass expansion may (A) impinge permission from P Cappabianca et al: Size does not matter. The intrigue of giant — FIGURE 392-5 Histology of prolactinoma. Microscopic view of lactotrope cells showing PRL secretion and characteristic morphology.
Figure 6¶

Caption: FIGURE 392-6 Large invasive prolactinoma successfully treated with cabergoline. A–B. invading the skull base. PRL level was 122,260 μg/L. Four days after cabergoline was tumor regression after 40 months of treatment, with PRL levels stable at 25 μg/L. Large prolactinoma. N Engl J Med 363:177, 2010.) increasing the dose. Most patients are controlled with a daily dose of <7.5 mg (2.5 mg tid). SIDE EFFECTS — FIGURE 392-6 MRI of craniopharyngioma. Sagittal T1-weighted image showing a cystic, calcified suprasellar mass derived from Rathke's pouch.
Figure 7¶

Caption: FIGURE 392-6 Large invasive prolactinoma successfully treated with cabergoline. A–B. invading the skull base. PRL level was 122,260 μg/L. Four days after cabergoline was tumor regression after 40 months of treatment, with PRL levels stable at 25 μg/L. Large prolactinoma. N Engl J Med 363:177, 2010.) increasing the dose. Most patients are controlled with a daily dose of <7.5 mg (2.5 mg tid). SIDE EFFECTS — FIGURE 392-7 MRI of pituitary metastasis. Axial T1-weighted image showing a lesion in the posterior pituitary consistent with metastatic deposit (e.g., from breast cancer).
Figure 8¶

Caption: FIGURE 392-6 Large invasive prolactinoma successfully treated with cabergoline. A–B. invading the skull base. PRL level was 122,260 μg/L. Four days after cabergoline was tumor regression after 40 months of treatment, with PRL levels stable at 25 μg/L. Large prolactinoma. N Engl J Med 363:177, 2010.) increasing the dose. Most patients are controlled with a daily dose of <7.5 mg (2.5 mg tid). SIDE EFFECTS — FIGURE 392-8 MRI of stalk compression. Coronal T1-weighted image showing thickening of the pituitary stalk due to compression by a nonfunctioning adenoma.
Figure 9¶

Caption: FIGURE 392-6 Large invasive prolactinoma successfully treated with cabergoline. A–B. invading the skull base. PRL level was 122,260 μg/L. Four days after cabergoline was tumor regression after 40 months of treatment, with PRL levels stable at 25 μg/L. Large prolactinoma. N Engl J Med 363:177, 2010.) increasing the dose. Most patients are controlled with a daily dose of <7.5 mg (2.5 mg tid). SIDE EFFECTS — FIGURE 392-9 MRI of hypothalamic hamartoma. Axial T2-weighted image showing a lesion contiguous with the pituitary stalk and hypothalamus, often associated with precocious puberty.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.