Systemic Lupus Erythematosus¶
Chapter 368 | Part 11: Immune-Mediated, Inflammatory, and Rheumatologic Disorders · Part 11 – Rheumatology & Immunology
Detailed clinical reference synthesised from Harrison's Principles of Internal Medicine, 22nd Edition
🔑 Key Clinical Points¶
- SLE is defined as an autoimmune disease in which overactive innate and adaptive immune systems cause tissue damage through the effects of autoantibodies and immune complexes.
- ANA titer of at least 1:80 is required for EULAR/ACR classification criteria; repeated negative tests by immunofluorescence make a diagnosis unlikely.
- Anti-dsDNA antibodies correlate with disease activity and nephritis; anti-Sm is the most specific autoantibody for SLE.
- Hydroxychloroquine (HCQ) is first-line therapy for skin manifestations and arthritis; dose is 5 mg/kg per day (actual body weight) with retinal monitoring.
- Lupus nephritis classification is based on renal biopsy (ISN/RPS criteria); aggressive therapy is reserved for class III (focal proliferative), class IV (diffuse proliferative), or class V (membranous).
- Type 1 lupus has classic findings (nephritis, arthritis) and responds to immunosuppression; Type 2 lupus has predominant symptoms of fatigue, brain fog, and depression and is less responsive to therapy.
- Drug-induced lupus is characterized by antihistone antibodies and is often abrogated by discontinuation of the offending drug.
- Stroke has been reported in up to 19% of patients with SLE, a result of atherosclerosis and increased risk from antiphospholipid syndrome (APS).
- Approximately 90% of affected individuals are women, most of childbearing age; prevalence is nine times higher in women compared to men.
- HCQ use during pregnancy and breastfeeding has been demonstrated to be safe; risk factors for retinal toxicity include higher dose and longer duration of use.
📑 Table of Contents¶
- 1. DEFINITION & OVERVIEW
- 1.1 Iatrogenic Autoimmunity
- 2. EPIDEMIOLOGY
- 3. ETIOLOGY & PATHOPHYSIOLOGY
- 3.1 Genetics
- 3.2 Innate Immunity
- 3.3 Adaptive Immunity
- 4. CLINICAL FEATURES
- 4.1 Musculoskeletal Manifestations
- 4.2 Cutaneous Manifestations
- 4.3 Renal Manifestations
- 4.4 Nervous System Manifestations
- 4.5 Vascular, Cardiac, and Pulmonary Manifestations
- 4.6 Hematologic Manifestations
- 4.7 Gastrointestinal and Ocular Manifestations
- 5. DIFFERENTIAL DIAGNOSIS
- 6. INVESTIGATIONS & DIAGNOSIS
- 6.1 Diagnostic Criteria
- 6.2 Laboratory Testing
- 7. MANAGEMENT & TREATMENT
- 7.1 General Measures and Antimalarials
- 7.2 Treatment Algorithm
- 8. PROGNOSIS & COMPLICATIONS
- 9. SPECIAL CONSIDERATIONS
- 10. KEY PEARLS & CLINICAL TRAPS
- Flowcharts & Algorithms
- Figures & Illustrations
📋 Figures in This Chapter¶
1. DEFINITION & OVERVIEW¶
Systemic lupus erythematosus (SLE) is an autoimmune disease in which overactive innate and adaptive immune systems cause tissue damage through the effects of autoantibodies and immune complexes. Many organ systems can be affected, with cutaneous, musculoskeletal, and renal systems most involved, followed by pulmonary, hematologic, cardiovascular, serosal, and central nervous system involvement. Constitutional symptoms are often present. Autoantibodies can be detected years prior to a clinical diagnosis. Approximately 90% of affected individuals are women, most of childbearing age; however, the disease can also affect neonates and older children, men, and elderly individuals.
1.1 Iatrogenic Autoimmunity¶
Checkpoint Inhibitors Immune checkpoint inhibitors (ICIs) have revolutionized cancer treatment. They act by blocking inhibitory molecules involved in regulating T lymphocytes, promoting the destruction of tumors by the adaptive immune system. Immune-related adverse events (irAEs) have been described as toxic complications related to tissue-specific autoimmunity from the use of ICIs, and they can persist after withholding therapy and using immunomodulatory therapy to control them. Among these complications, diabetes mellitus, arthritis, thyroiditis, and colitis are prevalent. Up to 85% of individuals treated with an ICI may develop irAEs. Most of these are mild, and overall survival and time to malignancy progression are not influenced by the development of these immune complications.
Drug-Induced Autoimmunity In addition to ICIs, many drugs have been linked to the development of autoimmunity. These include antibiotics, antihypertensives, antiarrhythmics, TNF inhibitors, and antiseizure medications, among many others. Drug-induced lupus is a well-described entity, and patients developing this condition have characteristic detection of antihistone antibodies. In many cases, discontinuation of the drug may be sufficient for symptoms to be abrogated or decreased.
2. EPIDEMIOLOGY¶
The Centers for Disease Control and Prevention (CDC) National Lupus Registry estimates the prevalence in the United States to be 204,295 cases, with overall prevalence being nine times higher in women compared to men. The highest prevalence is seen in Black women, followed by Hispanic, White, and Asian/Pacific Islander women. Among men with SLE, Black men have the highest prevalence and White men have the lowest. Prognosis and survival from SLE can vary widely by geographic region, race, ethnicity, and access to both care and medications.
3. ETIOLOGY & PATHOPHYSIOLOGY¶
SLE reflects multiple immunoregulatory defects and is characterized by the production of autoantibodies to cellular components, especially nucleic acids and nuclear proteins, that promote inflammation and tissue damage. The interaction of environmental factors with the stochastic dysregulation of genes controlled by common genetic variations underpins the development of SLE. About a quarter of monozygotic twins are clinically concordant for SLE, implicating environmental factors in disease expression. Low socioeconomic status is associated with the development of SLE and may include exposures to environmental toxins, including mercury, pesticides, and diet. Viral infections such as Epstein-Barr virus likely contribute to disease development by inducing the IFN response during an acute infection or by expressing viral proteins that activate SLE susceptibility genes. Ultraviolet light can impact DNA methylation, generate self-stimulatory nucleic acids, activate keratinocyte immune responses, and produce autoreactive T cells. Emerging evidence suggests that the gut microbiome may increase gut permeability, promoting the translocation of certain gut microbes into the blood with the development of lupus-specific autoantibodies contributing to SLE pathogenesis.
3.1 Genetics¶
SLE has a strong genetic component as evidenced by familial clustering, a greater incidence in monozygotic twins versus dizygotic (24% vs 2%), and the identification of >150 susceptibility loci from genome-wide association studies (GWAS). Genetic studies have identified groups of immunoregulatory genes associated with SLE, reflecting common pathways that may contribute to pathogenesis. It is possible that each pathway differentially contributes to the expression of disease, accounting for clinical heterogeneity. The most common genetic association is within the major histocompatibility complex that contains genes for antigen presentation molecules (class I and class II), several complement components, and cytokines. Nearly half of the susceptibility loci are associated with type 1 interferon (IFN) production or its downstream signaling. For example, some susceptibility genes lead to increased IFN through defects in nucleic acid sensing and metabolism (TLR7, ADAR, IFIH1, SAMHD1, RNASEH2B, and TREX1) or nucleic acid degradation (DNASE1, DNASE1L3), whereas other IFN-related susceptibility genes amplify the IFN response (IRF5, IRF7, IRF8, STAT4). Many predisposing genes are immune cell type specific, affecting activation and survival of T cells (OX40L) or B cells (BANK1, BACH2).
3.2 Innate Immunity¶
Multiple innate immune cell defects contribute to increased cellular breakdown and reduced cell debris clearance, culminating in the production of autoantibodies. Neutrophils are typically short-lived and abundant immune cells, representing a large burden of cellular debris that is usually cleared without inducing an immune response. Patients with SLE, however, have neutrophils with higher turnover, delivering a large load of stimulatory nucleic acids and autoantigens. Cellular debris and immune complexes are typically cleared by macrophages and dendritic cells through complement-mediated phagocytosis and nucleic acid digestion; these mechanisms are dysregulated in SLE. In turn, the burden of cellular debris exceeds that which can be cleared in SLE, triggering nucleic acid sensors and toll-like receptors (TLRs) in plasmacytoid dendritic cells to express type 1 IFN. Consequently, IFN primes neutrophils for further turnover and sensitizes the adaptive immune system. Other innate immune cells with aberrant function in SLE include classical monocytes that infiltrate and repair injured tissue due to immune complex deposition and nonclassical monocytes that patrol the vascular lumen for injury. Sustained activation of these subsets from SLE leads to tissue inflammation and scarring.
3.3 Adaptive Immunity¶
B cells play a central role in the SLE pathogenesis based on the production of autoantibodies due to their loss of tolerance. Autoantibodies, especially those complexed with nucleic acid, are pathogenic mediators of tissue inflammation and damage in SLE. These autoantibodies induce type 1 IFN and other inflammatory mediators and activate patrolling monocytes in tissue. A subset of circulating age-associated B cells (ABCs) expands in SLE patients and eventually matures into autoantibody-secreting plasma cells, indicating that SLE patients have an increased population of B cells primed to generate pathogenic autoantibodies. Similar to B cells, T cells lose tolerance and play a central role in the autoimmune response. In particular, both T follicular and T peripheral helper cells promote B-cell differentiation into pathogenic, high-affinity, autoantibody-secreting plasma cells. T regulatory cells meant to maintain tolerance in both T and B cells are defective in SLE.
4. CLINICAL FEATURES¶
The initial presentation of SLE is variable and may present as nonspecific constitutional symptoms with or without single-organ or multiorgan involvement. Autoantibody presence is helpful in attributing nonspecific symptoms to SLE. Symptoms can be mild to severe at any time over the course of the disease, including at initial presentation. Patients with severe disease often have systemic symptoms such as fever, anorexia, and unintentional weight loss. Approximately 15% of patients have relatively mild disease, which may or may not be accompanied by fatigue, brain fog, and/or mild arthralgia. The majority of SLE patients have active disease despite therapy or have frequent flares of their disease requiring treatment adjustment. Goals for SLE treatment are low-level disease activity or remission on ongoing therapy.
Table 1 — Table 368-1 Clinical Manifestations of Systemic Lupus Erythematosus¶
| System | Manifestations |
|---|---|
| Constitutional | Fatigue, malaise, fever, weight loss, anorexia |
| Cutaneous | Generalized Photosensitivity, oral and nasal ulcers, alopecia |
| Acute cutaneous | Malar rash, maculopapular rash, bullous lupus, toxic epidermal necrolysis variant |
| Subacute cutaneous | Lupus |
| Chronic forms of cutaneous lupus | Discoid rash, panniculitis, tumid lupus, chilblains, verrucous lupus, cutaneous vasculitis |
| Musculoskeletal | Arthralgia, myalgia, polyarthritis, Jaccoud hand deformity, inflammatory myopathy, avascular necrosis of bone |
| Hematologic | Anemia, leukopenia, lymphopenia, thrombocytopenia, lymphadenopathy, splenomegaly, venous or arterial thrombosis, atypical hemolytic-uremic syndrome |
| Cardiopulmonary | Pleurisy, pericarditis, pleural and pericardial effusions, myocarditis, endocarditis, pneumonitis, coronary artery disease, interstitial fibrosis, pulmonary hypertension, diffuse alveolar hemorrhage, shrinking lung syndrome |
| Vascular | Pulmonary hypertension, stroke, myocardial infarction, peripheral arterial disease, pulmonary embolism, deep vein thrombosis, Raynaud's phenomenon |
| Neurologic | Cognitive dysfunction, mood disorder, depression, headache, seizures, mononeuropathy, polyneuropathy, stroke, transient ischemic attack, psychosis, aseptic meningitis, transverse myelitis |
| Renal | Proteinuria ≥500 mg/24 h, cellular casts, nephrotic syndrome, end-stage renal disease |
| Gastrointestinal | Nausea, abdominal pain, diarrhea, elevated liver enzymes, lupus peritonitis or enteritis, vasculitis, pancreatitis |
| Ocular | Sicca syndrome, conjunctivitis, episcleritis, scleritis, uveitis, retinal vasculitis |
4.1 Musculoskeletal Manifestations¶
Nonspecific arthralgia and myalgia are common in SLE. Lupus arthritis is characterized by a polyarthritis most commonly involving the wrists, the metacarpal-phalangeal and proximal interphalangeal joints of the hands, and the knees. In some cases, damage of the periarticular ligaments may lead to Jaccoud-like changes. Rheumatoid arthritis and SLE can occur simultaneously, and this overlap is denoted by the term rhupus. While radiographs rarely demonstrate erosive changes in lupus arthritis, emerging studies with magnetic resonance imaging (MRI) and ultrasound imaging suggest a possible erosive pattern that is less severe than that seen with rheumatoid arthritis. Pain in a single hip, shoulder, or knee out of proportion to other joints should prompt consideration of avascular necrosis of bone, particularly with history of corticosteroid usage. Inflammatory muscle disease is also seen in lupus, characterized by symmetrical proximal weakness, elevated creatine kinase and aldolase levels, and inflammatory changes on muscle biopsy. Corticosteroids used to treat SLE may cause myopathy, as can antimalarials, although this is rare.
4.2 Cutaneous Manifestations¶
Three major categories of SLE skin manifestations include acute cutaneous lupus erythematosus (ACLE), subacute cutaneous lupus erythematosus (SCLE), and chronic cutaneous lupus. ACLE occurs with SLE disease activity, whereas SCLE and certain forms of chronic cutaneous lupus may be seen in the absence of systemic disease. The most classic form of ACLE is the malar rash, which appears in a 'butterfly' distribution across the cheeks of the face with nasolabial sparing. Due to its photosensitive nature, slightly raised lesions, and location on the face, this may be confused with rosacea, which involves the nasolabial folds. ACLE may also present as a generalized maculopapular rash in sun-exposed areas of the body, bullous lupus, toxic epidermal necrolysis variant, and generalized photosensitivity. SCLE is a photosensitive rash associated with anti-Ro or SSA antibodies that is characterized by flat, red-rimmed annular or psoriasiform lesions. Discoid lesions are rough, circular lesions that are slightly raised, scaly, and hyperpigmented with depigmented, atrophic centers and erythematous rims, and are the most common chronic dermatitis seen in SLE. Other chronic forms of cutaneous lupus include hypertrophic or verrucous lupus, lupus panniculitis, tumid lupus, chilblains lupus, and discoid lupus/lichen planus overlap. Nonspecific cutaneous manifestations that may be seen in SLE include nonscarring alopecia, oral and nasal painful or nonpainful mucosal ulcers, and the less common leukocytoclastic and urticarial vasculitides. Approximately one-third of SLE patients also experience Raynaud's phenomenon, a condition in which small blood vessels undergo vasospasm with exposure to cold environment or stress, causing a classically triphasic color change of the fingers and/or toes, inducing white (blanching), blue (cyanosis), and red (hyperemia) coloring of the extremities.
4.3 Renal Manifestations¶
Nephritis occurs in ~35% of patients with SLE and is more severe in non-White patients, more frequently leading to end-stage renal disease (ESRD) despite therapy. Nephritis can be seen with or without extrarenal SLE disease and may be present in an asymptomatic patient. Patients should therefore be screened at regular 3-month intervals with a random urine protein-to-creatinine ratio (UPCr) or a 24-h urine protein and creatinine determination. Usually, nephritis is seen in patients with elevated serum anti–double-stranded DNA (dsDNA) antibody titers and decreased serum levels of C3 and/or C4, and trends of these biomarkers over time may serve as warning signs of impending active nephritis. Renal biopsy is generally prompted by a UPCr of >0.5 and/or declining kidney function. Classification is based on renal pathology from a renal biopsy and is defined by the International Society of Nephrology/Renal Pathology Society criteria. Aggressive therapy is generally reserved for patients with class III (focal proliferative glomerulonephritis), class IV (diffuse proliferative glomerulonephritis), or a combination of class III or IV with class V disease (membranous). Approximately 20% of lupus nephritis patients with significant proteinuria tend to have class V disease and more often have nephrotic syndrome. Patients with class V lupus nephritis have a better overall prognosis than patients with class III or IV lupus nephritis and are treated in the same manner as class III and IV. Patients who do not respond to treatment may require a repeat biopsy to evaluate for a change in class. In the United States, ~20% of SLE patients with class IV lupus nephritis die or develop ESRD within 10 years. Newer approaches are being developed to both increase renal survival and spare patients from accelerated atherosclerosis, hypertension, hyperglycemia, and hyperlipidemia, all of which can be accelerated by renal disease and corticosteroid usage.
4.4 Nervous System Manifestations¶
Lupus patients experience neuropsychiatric manifestations of disease that are characterized by diffuse or focal symptoms. The most common diffuse symptom is 'brain fog,' a cognitive dysfunction or slowing of thought. The etiology is not well understood but may be related to autoantibodies crossing the blood-brain barrier, leading to local inflammation and neuronal damage. Headache is common and often severe, although it may be difficult to distinguish from migraine or tension headache. Seizures of any type may be seen in neuropsychiatric lupus. Acute psychosis and confusional states are uncommon but can occur in SLE without any structural abnormalities. In these cases, glucocorticoid-induced psychosis and infectious encephalitis should be ruled out. Transverse myelitis is a rare SLE complication that manifests as bilateral neurologic deficits such as symmetric motor weakness and decreased sensation due to spinal cord inflammation.
4.5 Vascular, Cardiac, and Pulmonary Manifestations¶
Coronary heart disease from accelerated atherosclerosis in SLE is a significant cause of morbidity and premature death, especially in young patients without other risk factors. Vascular injury due to excessive type 1 IFN and cellular death may predispose to atherosclerosis. The risk of coronary artery disease and vascular thrombosis is further increased in SLE patients with antiphospholipid syndrome (APS), an autoimmune syndrome that commonly occurs in SLE and is characterized by arterial, venous, or small vessel thromboembolic events in the presence of antiphospholipid antibodies with prothrombotic properties. Stroke has been reported in up to 19% of patients with SLE, a result of atherosclerosis and increased risk from APS. In addition to stroke, APS appears to be a significant risk factor for other focal central nervous system manifestations including seizures and transverse myelitis. Pulmonary arterial hypertension and pulmonary embolism may be present with or without APS. All layers of the heart may be involved in SLE, with pericardial involvement being the most frequent. Pericarditis can typically be managed with anti-inflammatory medications, colchicine, and anti-IL-1–directed therapies, or without glucocorticoids, and rarely leads to tamponade physiology. Myocarditis is rarer and may lead to left-sided heart failure and/or arrhythmia. Libman-Sacks endocarditis, a fibrinous sterile form of endocarditis that may be associated with antiphospholipid antibodies, may increase the risk for embolic events. Right-sided heart failure may be present with pulmonary arterial hypertension or chronic lung disease. The most common pulmonary manifestation of SLE is pleuritis, which can occur with or without exudative pleural effusions. Acute pneumonitis may present in active SLE, with pulmonary infiltrates often appearing indistinguishable from infection on imaging. Diffuse alveolar hemorrhage with capillaritis and interstitial lung disease may also be seen. Restrictive lung defect from reduced lung volumes, also known as shrinking lung syndrome, is uncommon but may occur.
4.6 Hematologic Manifestations¶
Cytopenias in SLE are often multifactorial and may be associated with disease activity, medication use, or infection. Anemia is the most common hematologic manifestation of SLE and is present in >50% of SLE cases, with anemia of chronic disease representing approximately one-third of cases; other causes for anemia in SLE include iron deficiency, autoimmune hemolytic anemia, aplastic anemia, microangiopathic hemolytic anemia, and medication effect. Leukopenia (<4000/µL) is common, occurring in about half of SLE patients, and usually consists of lymphopenia (<1500/µL), rather than neutropenia. Thrombocytopenia (<150,000/µL) is usually mild but can also be severe (<50,000/µL) and pose significant bleeding risk. It is most commonly due to immune mechanisms and may be associated with APS or antiphospholipid antibodies against platelet antigens. Splenomegaly, splenic atrophy, and asplenism may all be seen in SLE. Depending on severity of the abnormal blood counts, treatment considerations include glucocorticoids, anti-CD20 biologic agents, platelet growth factors, intravenous immunoglobulin, and/or splenectomy in resistant cases. Rarely, atypical hemolytic-uremic syndrome (formerly designated as thrombotic thrombocytopenic purpura or the syndrome of microvascular thrombotic crisis) may occur. This is a condition that presents with hemolysis, thrombocytopenia, and microvascular thrombosis, as well as brain and other tissue involvement. Laboratory testing shows schistocytes in the peripheral blood smear, low levels of ADAMTS13 activity, and elevated serum lactate dehydrogenase levels.
4.7 Gastrointestinal and Ocular Manifestations¶
Lupus can involve any part of the gastrointestinal tract. Flaring SLE activity has been associated with nonspecific gastrointestinal symptoms including nausea, vomiting, and diarrhea. Abdominal pain may be caused by lupus peritonitis, enteritis, pancreatitis, or vasculitis. Mesenteric vasculitis can lead to intestinal perforation, bleeding, and ischemia and requires high doses of glucocorticoids for treatment. Impaired intestinal motility and protein-losing enteropathy may also occur. Elevated liver enzymes are common and may be associated with flaring disease activity, medications, and/or coexisting autoimmune hepatobiliary disease. Mesenteric thrombosis, Budd-Chiari syndrome, and hepatic venoocclusive disease can be observed with concomitant APS. Keratoconjunctivitis sicca is common in SLE, even in the absence of secondary Sjögren's syndrome. Retinal vasculitis, optic neuritis, uveitis, scleritis, peripheral ulcerative keratitis, episcleritis, and nonspecific conjunctivitis may all be seen. Retinal vasculitis is rare but, when present, requires aggressive immunosuppression to prevent blindness. Adverse ocular effects from medications include cataracts and glaucoma from glucocorticoid treatment and hydroxychloroquine (HCQ)-induced maculopathy.
5. DIFFERENTIAL DIAGNOSIS¶
For the diagnosis of SLE, an individualized approach should be taken based on available clinical data and excluding other disease entities. The hope is that better understanding of the pathophysiology of overt development of autoimmune diseases and their associated organ damage will lead to primary or secondary prevention strategies in conditions such as T1DM, RA, and SLE. One promising example is where infants at risk of developing multiple T1DM autoAbs and clinical disease are being studied to determine if certain interventions (oral insulin) can delay or stop progression to overt disease. Studies have also been performed on people considered at risk for RA to assess delayed disease progression, but more studies are needed to better characterize the utility of these preventive strategies. Overall, opportunities to initiate early therapy depend on the availability of serologic tests that can predict disease, to initiate treatments that have a good efficacy-to-safety ratio.
6. INVESTIGATIONS & DIAGNOSIS¶
An elevated titer of ANA reflects the underlying immune dysregulation in SLE. Measuring ANA presence and titer is the best screening test for SLE because ANAs are seen in almost all SLE patients. Repeated negative tests by immunofluorescence make a diagnosis unlikely, but rare cases of ANA-negative SLE do occur. Classification of SLE by current EULAR/ACR criteria requires the presence of an ANA titer of 1:80 for enrollment of patients into SLE clinical trials; however, the diagnosis is based on clinical and laboratory features and ultimately, clinical judgement. Numerous ANAs have been identified that target nuclear antigens; in particular, components of the nucleosome (DNA wrapped around a histone octamer) or RNA-binding proteins (RBPs). In SLE, the most common antinucleosome antibodies are anti-dsDNA antibodies that are present in ~40–60% of patients and highly specific (75–99%). Anti-Smith (anti-Sm) antibodies are the most common anti-RBP, present in ~30% of patients, and are highly specific for SLE (55–100%). Given their high specificity, both anti-dsDNA and anti-Sm antibodies are the only ANA subtypes that are included in the current EULAR/ACR criteria. Anti-Ro (SS-A) antibodies, which recognize a protein complexed primarily to 60-kDa and 52-kDa RNAs, are often seen in ANA-negative SLE patients and are associated with risk for neonatal lupus, sicca symptoms, photosensitivity, and SCLE. Antihistone antibodies may be associated with drug-induced lupus. Antiphospholipid antibodies, as measured by IgG, IgM, and IgA isotypes of anticardiolipin and anti–beta-2 glycoprotein-1 antibodies (anti–βGP-1), and the lupus anticoagulant usually obtained using the dilute Russell venom viper time (dRVVT) are present in ~50% of SLE patients. These antibodies, especially the IgG and IgA isotypes of cardiolipin and βGP-1, and the lupus anticoagulant are associated with arterial and venous thrombosis, thrombocytopenia, and recurrent fetal loss.
6.1 Diagnostic Criteria¶
Currently, the 2019 European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria for SLE and the 2012 Systemic Lupus International Collaborating Clinics (SLICC) SLE classification criteria are both utilized for enrollment of patients into clinical trials. For the EULAR/ACR criteria, an antinuclear antibody (ANA) titer of at least 1:80 is required. Clinical criteria are then weighed by points within several clinical domains, and a patient requires at least 10 points to meet criteria. No single clinical or laboratory value leads to 10 points except for a renal biopsy revealing class III or IV lupus nephritis. Certain subsets of patients with SLE are currently not included in clinical trials, including patients with incomplete lupus or features of SLE without an established diagnosis and the ~5% of SLE patients who are ANA negative. Researchers have suggested that if predictive or diagnostic biomarkers for progression are developed, then early treatment for patients with incomplete lupus could prevent progression to SLE. Furthermore, because the EULAR/ACR criteria exclude ANA-negative patients from participating in clinical trials, the opportunity to understand whether new drugs have a role in this disease subset is also underserved.
Table 2 — Table 368-2 Autoantibodies in Systemic Lupus Erythematosus¶
| Antibody | Prevalence (%) | Clinical Associations |
|---|---|---|
| ANA | 95 | Titers of 1:80 or higher are considered positive and are sensitive for SLE but not specific. |
| Anti-dsDNA | 70 | Higher titers are more specific for SLE. Levels correlate with disease activity and nephritis. |
| Anti-Sm | 30 | Most specific autoantibody for SLE and more common in black and Asian patients. May correlate with CNS manifestations and incidence of nephritis. |
| Anti-Ro (SS-A) | 30 | Not SLE-specific. Associated with sicca syndrome, neonatal lupus and congenital heart block, and subacute cutaneous lupus. |
| Antiribosomal P | 15 | May be associated with CNS involvement. Specific for SLE but not sensitive. |
| Anti–U1-RNP | 13 | Associated with musculoskeletal and lung impairment. |
| Antiphospholipid | 50 | Arterial or venous thrombosis, pregnancy morbidity and fetal loss, thrombocytopenia. Associated with CNS manifestations, Libman-Sacks endocarditis, hypertension, pulmonary hypertension. |
| Anti–β2-glycoprotein | 2 | Associated with CNS manifestations, Libman-Sacks endocarditis, hypertension, pulmonary hypertension. |
6.2 Laboratory Testing¶
Laboratory testing for the diagnosis of SLE includes immunologic, renal, and hematologic domains. Among the immunologic domain, elevated titers of ANA confer a high likelihood of SLE. Although ANAs are present at low levels in healthy patients, the threshold for a positive test of 1:80 or higher favors SLE. In the setting of ANA negativity with a clinical presentation highly suggestive of SLE, anti-Ro (SSA) confers a high likelihood of SLE. Further immunologic testing for anti-dsDNA, anti-Sm, anti-Ro, anti-La, antiribosomal P, antiphospholipid antibodies, and anticardiolipin is necessary. Laboratory testing is required at initial presentation to screen for hematologic abnormalities including leukopenia, lymphopenia, thrombocytopenia, and anemia of chronic inflammation, hemolytic anemia, and atypical hemolytic-uremic syndrome. Initial testing for renal abnormalities is necessary to determine an estimated glomerular filtration rate (eGFR), serum creatinine, and either a random or 24-h UPCr. When commercially available, consideration should be given to measuring urinary biomarkers recently noted to be associated with renal inflammatory and fibrotic disease, including urinary interleukin 16, CD163, type 1 IFN, and transforming growth factor-beta (TGF–β). Testing for eGFR, serum creatinine, and a random or 24-h UPCr level is essential for routine follow-up care with consideration of a renal biopsy when urinary protein exceeds a new level of UPCr of 0.5 to rule out active lupus nephritis. Consideration should be given to measuring urinary biomarkers on a quarterly basis when available.
7. MANAGEMENT & TREATMENT¶
EULAR recently updated recommendations for the management of SLE and lupus nephritis. Goals for SLE treatment include improved disease activity with minimal symptoms, prevention of damage, and improved quality of life. Adherence to treatment plans is essential. Achieving these goals with safe treatments and minimal corticosteroids is an integral aspect of achieving low levels of disease activity and remission. If corticosteroids are utilized for active or flaring disease, the goal should be to utilize the lowest possible dose to suppress disease activity. Aggressive therapy should be reserved for life-threatening manifestations or those that could lead to damage. Consideration should be given to minimize both the effects of active disease and complications from treatment. Renal treatment goals based on EULAR recommendations include (1) a goal that all patients should at least achieve a state of low disease activity, defined by a Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score of 0–4; or (2) remission, defined as a SLEDAI score of 0, both with stable use of HCQ, immunosuppressives or biologics, and a daily dose of 5 mg of prednisone (or glucocorticoid equivalent) or less.
7.1 General Measures and Antimalarials¶
All SLE patients should use daily sunscreen on sun-exposed areas, maintain a healthy diet and weight, avoid smoking, exercise regularly, maintain compliance with vaccination recommendations, and regularly monitor and achieve control of their blood pressure, lipid, and glucose levels. Antimalarials should be used in all patients unless contraindicated, as they have been shown to prevent flares, increase overall survival, and decrease the risk of developing renal disease and accelerated atherosclerosis, among other benefits. HCQ, one of the first drugs approved by the U.S. Food and Drug Administration (FDA) for use in SLE, is the most common antimalarial used and is first-line therapy for patients with skin manifestations and arthritis. It is the most useful drug to improve fatigue in SLE. Use during pregnancy and during breastfeeding has been demonstrated to be safe. Appropriate dosing of HCQ is under investigation, as higher doses may be associated with better disease control. Current recommendations are for dosing up to 5 mg/kg per day (actual body weight) with performance of retinal evaluations at baseline and at yearly intervals to monitor for macular effects, which are associated with prolonged usage. Risk factors for retinal toxicity from HCQ include higher dose and longer duration of use; however, higher doses may also confer better disease control, and overall risk for maculopathy remains low. The most appropriate dose of HCQ for SLE treatment thus remains unclear. Other side effects of HCQ may include rash, nausea, vomiting and diarrhea, and skin dyspigmentation. Tobacco use may interfere with the efficacy of antimalarials. Alternative antimalarials include chloroquine, which has a higher risk of retinal toxicity, and quinacrine, which has a higher risk of retinal toxicity.
7.2 Treatment Algorithm¶
Treatment of Non-Renal Systemic Lupus Erythematosus is stratified by severity. Mild disease includes constitutional symptoms; mild arthritis; rash ≤9% body surface area; platelet count (PLTs) 50–100 × 109/L; SLEDAI ≤6; BILAG C or ≤1 BILAG B manifestation. Moderate disease includes moderate-to-severe arthritis ('rheumatoid arthritis–like'; rash 9–18% body surface area [BSA]; PLTs 20–50 × 109/L; serositis; SLEDAI 7–12; ≥2 BILAG B manifestations). Severe disease includes major organ-threatening disease (e.g., cerebritis, myelitis, pneumonitis, mesenteric vasculitis); thrombocytopenia with platelets 18% BSA; SLEDAI >12; ≥1 BILAG A manifestation. First-line therapy for mild disease includes HCQ (all patients unless contraindicated), sun protection, exercise, no smoking, balanced diet, normal body weight, blood pressure/lipid/glucose control, and acetylsalicylic acid (in aPL+/APS). Second-line therapy for mild disease includes methotrexate (MTX), azathioprine (AZA), mycophenolate mofetil (MMF), and anifrolumab (ANI) or belimumab (BEL). First-line therapy for moderate disease includes HCQ, glucocorticoids (GC) ≤5 mg/day, MTX, AZA, MMF, and anifrolumab (ANI) or belimumab (BEL). Second-line therapy for moderate disease includes cyclophosphamide (CYC), rituximab (RTX), and CNI. First-line therapy for severe disease includes HCQ, GC ≤5 mg/day, MMF, ANI, BEL, and CNI. Second-line therapy for severe disease includes RTX, CNI at stable, tolerated dose, and immunosuppressive or biological agents. Grade A, B, C, D refer to recommendation strength.
8. PROGNOSIS & COMPLICATIONS¶
In the United States, ~20% of SLE patients with class IV lupus nephritis die or develop ESRD within 10 years. Newer approaches are being developed to both increase renal survival and spare patients from accelerated atherosclerosis, hypertension, hyperglycemia, and hyperlipidemia, all of which can be accelerated by renal disease and corticosteroid usage. Prognosis and survival from SLE can vary widely by geographic region, race, ethnicity, and access to both care and medications. Cancers commonly associated with autoimmunity include melanoma, thyroid cancer, and non-small-cell lung cancer. One specific example is the development of encephalitis through generation of Abs to N-methyl-d-aspartate glutamate receptor in women affected by ovarian cancer. In some cases, these events may be associated with favorable disease outcomes that may suggest a beneficial effect from the autoimmune response. The autoimmune manifestations can be severe, and management of these patients is difficult given the coexistent malignancy.
9. SPECIAL CONSIDERATIONS¶
Certain subsets of patients with SLE are currently not included in clinical trials, including patients with incomplete lupus or features of SLE without an established diagnosis and the ~5% of SLE patients who are ANA negative. Researchers have suggested that if predictive or diagnostic biomarkers for progression are developed, then early treatment for patients with incomplete lupus could prevent progression to SLE. Furthermore, because the EULAR/ACR criteria exclude ANA-negative patients from participating in clinical trials, the opportunity to understand whether new drugs have a role in this disease subset is also underserved. Drug-induced lupus is a well-described entity, and patients developing this condition have characteristic detection of antihistone antibodies. In many cases, discontinuation of the drug may be sufficient for symptoms to be abrogated or decreased. Immune checkpoint inhibitors (ICIs) have revolutionized cancer treatment. They act by blocking inhibitory molecules involved in regulating T lymphocytes, promoting the destruction of tumors by the adaptive immune system. Immune-related adverse events (irAEs) have been described as toxic complications related to tissue-specific autoimmunity from the use of ICIs, and they can persist after withholding therapy and using immunomodulatory therapy to control them. Among these complications, diabetes mellitus, arthritis, thyroiditis, and colitis are prevalent. Up to 85% of individuals treated with an ICI may develop irAEs. Most of these are mild, and overall survival and time to malignancy progression are not influenced by the development of these immune complications.
10. KEY PEARLS & CLINICAL TRAPS¶
Repeated negative tests by immunofluorescence make a diagnosis unlikely, but rare cases of ANA-negative SLE do occur. The most classic form of ACLE is the malar rash, which appears in a 'butterfly' distribution across the cheeks of the face with nasolabial sparing. Discoid lesions are rough, circular lesions that are slightly raised, scaly, and hyperpigmented with depigmented, atrophic centers and erythematous rims, and are the most common chronic dermatitis seen in SLE. Pain in a single hip, shoulder, or knee out of proportion to other joints should prompt consideration of avascular necrosis of bone, particularly with history of corticosteroid usage. Stroke has been reported in up to 19% of patients with SLE, a result of atherosclerosis and increased risk from APS. Approximately 90% of affected individuals are women, most of childbearing age; however, the disease can also affect neonates and older children, men, and elderly individuals. The highest prevalence is seen in Black women, followed by Hispanic, White, and Asian/Pacific Islander women. Among men with SLE, Black men have the highest prevalence and White men have the lowest.
Flowcharts & Algorithms¶
Reproduced from Harrison's 22nd Edition.
Flowchart 1¶

Caption: FIGURE 368-4 Treatment of nonrenal systemic lupus erythematosus (SLE). Notes: are equal options for second-line therapy in mild disease or first-line therapy in surface area; platelet count (PLTs) 50–100 × 109/L; SLEDAI ≤6; BILAG C or ≤1 BILAG arthritis–like”; rash 9–18% body surface area [BSA]; PLTs 20–50 × 109/L; serositis; disease (e.g., cerebritis, myelitis, pneumonitis, mesenteric vasculitis); or acute hemophagocytic syndrome; rash >18% BSA; SLEDAI >12; ≥1 BILAG A severe disease refers to cases of extrarenal SLE with nonmajor organ involvement but therapy in severe disease refers mainly to severe skin disease. For patients with severe anifrolumab; aPL, antiphospholipid antibodies; APS, antiphospholipid syndrome; AZA, calcineurin inhibitor; CYC, cyclophosphamide; GC, glucocorticoids; HCQ, RTX, rituximab; SLEDAI, SLE Disease Activity Index; VKA, vitamin K antagonists.
Flowchart 2¶
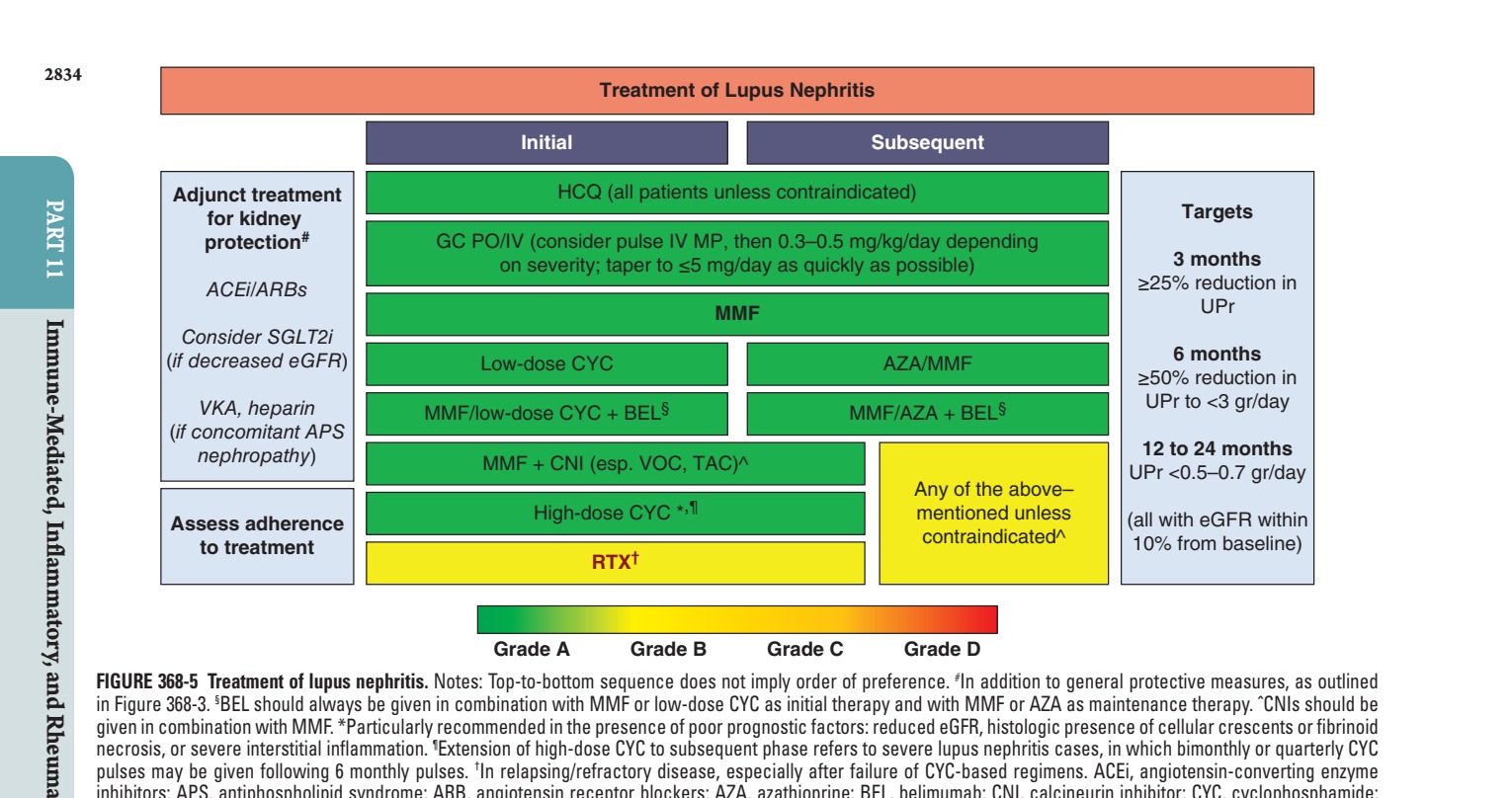
Caption: FIGURE 368-5 Treatment of lupus nephritis. Notes: Top-to-bottom sequence does not in Figure 368-3. §BEL should always be given in combination with MMF or low-dose CYC given in combination with MMF. *Particularly recommended in the presence of poor necrosis, or severe interstitial inflammation. ¶Extension of high-dose CYC to subsequent pulses may be given following 6 monthly pulses. †In relapsing/refractory disease, inhibitors; APS, antiphospholipid syndrome; ARB, angiotensin receptor blockers; AZA, eGFR, estimated glomerular filtration rate; GC, glucocorticoids; HCQ, hydroxychloroquine; os; RTX, rituximab; SGLT2i, sodium-glucose cotransporter 2 inhibitors; TAC, tacrolimus; permission from A Fanouriakis et al: Ann Rheum Dis 83:15, 2024.)
Figures & Illustrations¶
Reproduced from Harrison's 22nd Edition.
Figure 1¶

Caption: FIGURE 368-1 Pathogenesis of systemic lupus erythematosus (SLE). Autoantigens regulated by the innate immune system in SLE. Consequently, these autoantigens plasmacytoid dendritic cells (DCs), producing type 1 IFN, sensitizing the innate and to B-cell stimulation and the production of immune complexes against these lead to inflammation and repair and, eventually, fibrosis after recurrent SLE flares. EBV, — Figure 368-1: Pathogenesis of systemic lupus erythematosus (SLE). Autoantigens from neutrophil NETosis and cellular turnover overwhelm clearance mechanisms regulated by the innate immune system. Nucleic acid activates TLR signaling in plasmacytoid dendritic cells (DCs), producing type 1 IFN, sensitizing the innate and adaptive immune responses. DCs present autoantigens to autoreactive T cells, leading to B-cell stimulation and the production of immune complexes against these autoantigens.
Figure 2¶
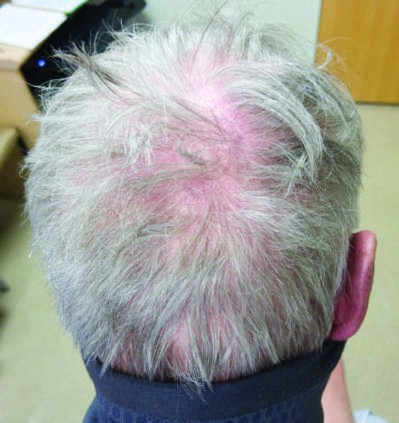
Caption: FIGURE 368-2 Characteristic musculoskeletal and cutaneous manifestations seen in chronic, nonerosive, and reversible deviations caused by joint capsule inflammation Patchy, nonscarring alopecia. C. Patchy scarring alopecia. D. Patchy alopecia rash. F. Subacute cutaneous lupus erythematosus (SCLE) with widespread, nonscarring Werth, MD. Panels A and F reproduced with permission from DoQuyen Huynh, MD.) — Figure 368-2A: Jaccoud arthropathy, characterized by chronic, nonerosive, and reversible deviations caused by joint capsule inflammation with subsequent fibrotic retraction and metacarpophalangeal joint subluxation.
Figure 3¶

Caption: FIGURE 368-2 Characteristic musculoskeletal and cutaneous manifestations seen in chronic, nonerosive, and reversible deviations caused by joint capsule inflammation Patchy, nonscarring alopecia. C. Patchy scarring alopecia. D. Patchy alopecia rash. F. Subacute cutaneous lupus erythematosus (SCLE) with widespread, nonscarring Werth, MD. Panels A and F reproduced with permission from DoQuyen Huynh, MD.) — Figure 368-2B: Patchy, nonscarring alopecia seen in systemic lupus erythematosus (SLE).
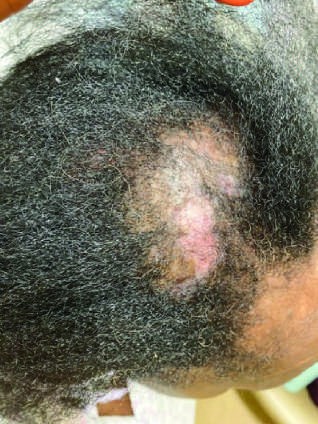
Figure 4¶

Caption: FIGURE 368-2 Characteristic musculoskeletal and cutaneous manifestations seen in chronic, nonerosive, and reversible deviations caused by joint capsule inflammation Patchy, nonscarring alopecia. C. Patchy scarring alopecia. D. Patchy alopecia rash. F. Subacute cutaneous lupus erythematosus (SCLE) with widespread, nonscarring Werth, MD. Panels A and F reproduced with permission from DoQuyen Huynh, MD.) — Figure 368-2C: Patchy scarring alopecia seen in systemic lupus erythematosus (SLE).
Figure 5¶
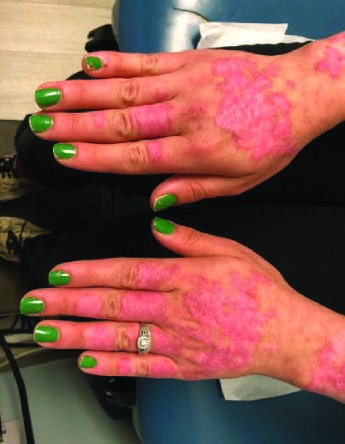
Caption: FIGURE 368-2 Characteristic musculoskeletal and cutaneous manifestations seen in chronic, nonerosive, and reversible deviations caused by joint capsule inflammation Patchy, nonscarring alopecia. C. Patchy scarring alopecia. D. Patchy alopecia rash. F. Subacute cutaneous lupus erythematosus (SCLE) with widespread, nonscarring Werth, MD. Panels A and F reproduced with permission from DoQuyen Huynh, MD.) — Figure 368-2D: Patchy alopecia associated with chronic, scarring discoid lupus changes seen in systemic lupus erythematosus (SLE).
Figure 6¶
Caption: FIGURE 368-2 Characteristic musculoskeletal and cutaneous manifestations seen in chronic, nonerosive, and reversible deviations caused by joint capsule inflammation Patchy, nonscarring alopecia. C. Patchy scarring alopecia. D. Patchy alopecia rash. F. Subacute cutaneous lupus erythematosus (SCLE) with widespread, nonscarring Werth, MD. Panels A and F reproduced with permission from DoQuyen Huynh, MD.) — Figure 368-2E: Classic photosensitive malar rash appearing in a 'butterfly' distribution across the cheeks of the face with nasolabial sparing.
Figure 7¶
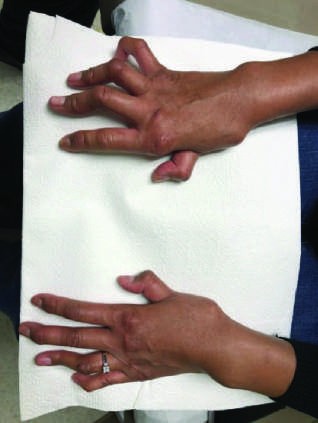
Caption: FIGURE 368-2 Characteristic musculoskeletal and cutaneous manifestations seen in chronic, nonerosive, and reversible deviations caused by joint capsule inflammation Patchy, nonscarring alopecia. C. Patchy scarring alopecia. D. Patchy alopecia rash. F. Subacute cutaneous lupus erythematosus (SCLE) with widespread, nonscarring Werth, MD. Panels A and F reproduced with permission from DoQuyen Huynh, MD.) — Figure 368-2F: Subacute cutaneous lupus erythematosus (SCLE) with widespread, nonscarring photosensitive features.
Figure 8¶

Caption: Figure 368-3: Ultrastructural features of lupus glomerulonephritis. Class I: Mesangial immune deposits without mesangial cell hypercellularity. Class II: Mesangial immune deposits with mesangial cell hypercellularity. Class III/IV: Subendothelial immune deposits with infiltration of mesangial and capillary leukocytes. Class V: Subepithelial immune deposits, no leukocyte infiltration.
Figure 9¶

Caption: FIGURE 368-3 Ultrastructural features of lupus glomerulonephritis. Class I: Mesangial immune deposits (black) without mesangial cell (red) infiltration. Class II: Mesangial immune deposits with mesangial cell hypercellularity, no leukocyte infiltration. Class III/IV: Subendothelial immune microscopy, with infiltration of mesangial and capillary leukocytes (dark green neutrophils and light green monocytes/macrophages). Class III/IV + V: with subepithelial and subendothelial immune deposits. Class V: Subepithelial immune deposits, no leukocyte infiltration. (Reproduced with permission al: Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: Clarification of definitions, and modified of Health activity and chronicity indices. Kidney Int 93:789, 2018, Figure 2.) — Figure 368-4: Treatment of nonrenal systemic lupus erythematosus (SLE). Top-to-bottom sequence does not imply order of preference; mild disease includes constitutional symptoms and mild arthritis; moderate disease includes moderate-to-severe arthritis and rash 9–18% body surface area; severe disease includes major organ-threatening disease and SLEDAI >12.
Generated from Harrison's Principles of Internal Medicine, 22nd Edition.